FastDTLmapper
Genome-wide gene gain/loss mapping tool using DTL(Duplication-Transfer-Loss) reconciliation method
Install / Use
/learn @moshi4/FastDTLmapperREADME
FastDTLmapper: Fast genome-wide DTL event mapper

![]()
![]()
Table of contents
Overview
Gene gain/loss is considered to be one of the most important evolutionary processes
driving adaptive evolution, but it remains largely unexplored.
Therefore, to investigate the relationship between gene gain/loss and adaptive evolution
in the evolutionary process of organisms, I developed a software pipeline FastDTLmapper
which automatically estimates and maps genome-wide gene gain/loss.
FastDTLmapper takes two inputs, 1. Species tree (Newick format) & 2. Genomic Protein CDSs (Fasta|Genbank format),
and performs genome-wide mapping of DTL(Duplication-Transfer-Loss) events by
DTL reconciliation of species tree and gene trees.
Additionally, FastDTLmapper can perform
Plot Gain/Loss Map Figure and
Functional Analysis (GOEA)
using packaged subtools.
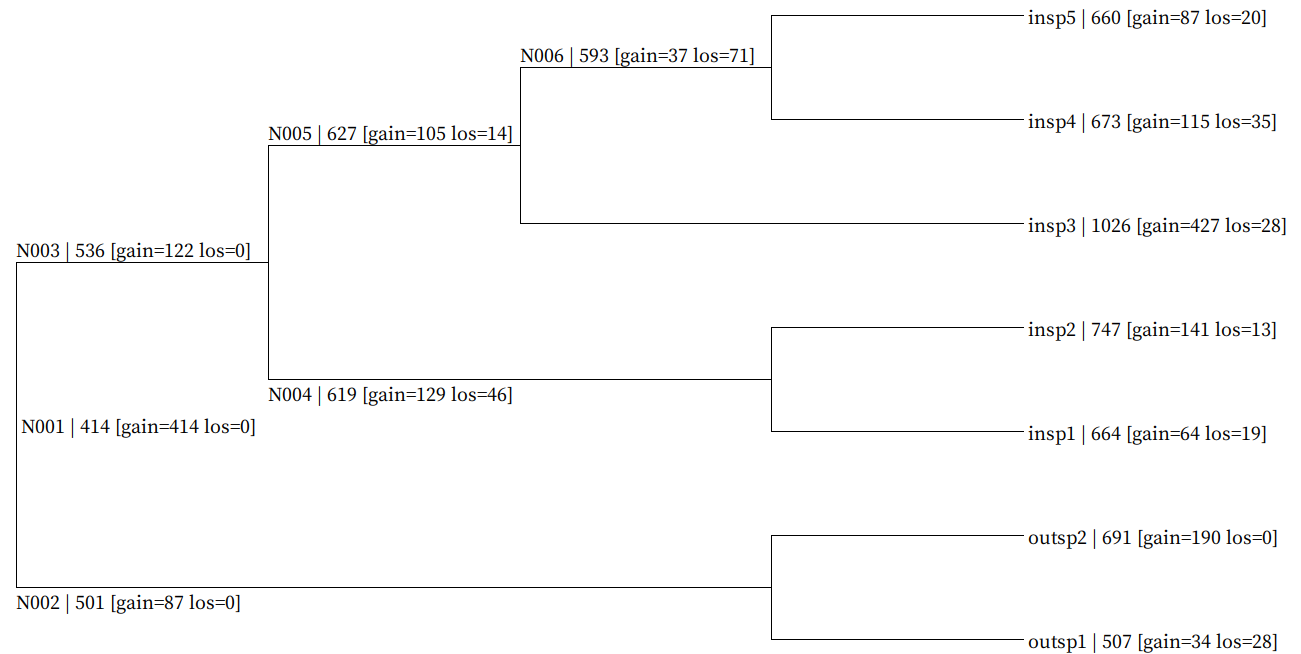
Fig. Genome-wide gain/loss map result example (all_gain_loss_map.nwk)
Each node gain/loss data is mapped in following format (NodeID | GeneNum [gain=GainNum los=LossNum])
Map data is embeded in newick format bootstrap value field and user can visualize using SeaView.
Install
FastDTLmapper is implemented in Python3(>=3.7) and runs on Linux (Tested on Ubuntu20.04).
:warning: Additionally, dependent tools require Python2.7 and Perl5. Since FastDTLmapper's dependencies are complex, it is recommended to use Docker image.
Install PyPI stable package:
pip install fastdtlmapper
Install latest development package:
pip install git+git://github.com/moshi4/FastDTLmapper.git
Use Docker (Image Registry):
docker pull ghcr.io/moshi4/fastdtlmapper:latest
docker run -it --rm ghcr.io/moshi4/fastdtlmapper:latest FastDTLmapper -h
Dependencies
Python package dependencies list here (auto installed with pip).
Well known python package numpy, pandas, scipy and
- BioPython
Utility tools for computational molecular biology - GOAtools
GOEA(GO Enrichment Analysis) tool - ETE3
Tree analysis and visualization tool
Following dependencies are packaged in src/fastdtlmapper/bin directory.
- OrthoFinder [v2.5.2]
Orthology inference tool - mafft [v7.487]
Sequences alignment tool - trimal [v1.4]
Alignment sequences trim tool - IQ-TREE [v2.1.3]
Phylogenetic tree reconstruction tool - Treerecs [v1.2]
Multifurcated gene tree correction tool - AnGST
DTL reconciliation tool (Requires Python 2.7 to run) - parallel [v20200922]
Job parallelization tool (Requires Perl5 to run)
BioPython:
Cock, P.J.A. et al.
Biopython: freely available Python tools for computational molecular biology and bioinformatics. (2009)
Bioinformatics 25(11) 1422-3
GOAtools:
Klopfenstein DV, Zhang L, Pedersen BS, ... Tang H
GOATOOLS: A Python library for Gene Ontologyy analyses (2018)
Scientific reports 8:10872
ETE:
Huerta-Cepas J., Serra F. and Bork P.
ETE 3: Reconstruction, analysis and visualization of phylogenomic data (2016)
Mol Biol Evol 33(6) 1635-1638
OrthoFinder:
Emms D.M. & Kelly S.
OrthoFinder: phylogenetic orthology inference for comparative genomics (2019)
Genome Biology 20:238
MAFFT:
Yamada, Tomii, Katoh.
Application of the MAFFT sequence alignment program to large data—reexamination of the usefulness of chained guide trees. (2016)
Bioinformatics 32:3246-3251
trimAl:
Salvador Capella-Gutierrez; Jose M. Silla-Martinez; Toni Gabaldon.
trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. (2009)
Bioinformatics 25: 1972-1973.
IQ-TREE:
B.Q. Minh, H.A. Schmidt, O. Chernomor, D. Schrempf, M.D. Woodhams, A. von Haeseler, R. Lanfear.
IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. (2020)
Mol. Biol. Evol. 37:1530-1534.
Treerecs:
Comte N, Morel B, Hasic D, Guéguen L, Boussau B, Daubin V, Penel S, Scornavacca C, Gouy M, Stamatakis A, et al.
Treerecs: an integrated phylogenetic tool, from sequences to reconciliations (2020)
Bioinformatics 36:4822–4824
AnGST:
Lawrence A David and Eric J Alm.
Rapid evolutionary innovation during an Archaean genetic expansion. (2010)
Nature. 469(7328):93-6
parallel:
</details>O. Tange
GNU Parallel - The Command-Line Power Tool, ;login: (2011)
The USENIX Magazine, February 2011:42-47.
Analysis Pipeline
This is brief description of analysis pipeline. See wiki for details.
- Grouping ortholog sequences using OrthoFinder
- Align each OG(Ortholog Group) sequences using mafft
- Trim each OG alignment using trimal
- Reconstruct each OG gene tree using IQ-TREE
- Correct each OG gene tree multifurcation using Treerecs
- DTL reconciliation of species tree & each OG gene tree using AnGST
- Aggregate and map genome-wide DTL reconciliation result
Command Usage
Basic Command
FastDTLmapper -i [fasta|genbank directory] -t [species tree file] -o [output directory]
Options
-i IN, --indir IN Input Fasta(*.fa|*.faa|*.fasta), Genbank(*.gb|*.gbk|*.gbff) directory
-t TREE, --tree TREE Input rooted species newick tree file
-o OUT, --outdir OUT Output directory
-p , --process_num Number of processor (Default: MaxProcessor - 1)
--dup_cost Duplication event cost (Default: 2)
--los_cost Loss event cost (Default: 1)
--trn_cost Transfer event cost (Default: 3)
--inflation OrthoFinder MCL inflation parameter (Default: 3.0)
--timetree Use species tree as timetree in AnGST (Default: off)
--rseed Number of random seed (Default: 0)
-v, --version Print version information
-h, --help Show this help message and exit
-
Timetree Option
If user set this option, input species tree must be ultrametric tree.
--timetree enable AnGST timetree option below (See AnGST manual for details).If the branch lengths on the provided species tree represent times, AnGST can restrict the set of possible inferred gene transfers to only those between contemporaneous lineages
-
Input Limitation
fasta or genbank files (--indir option)
:warning: Following characters cannot be included in file name '_', '-', '|', '.', '$'
species tree file (--tree option)
:warning: Species name in species tree must match fasta or genbank file name
Example Command
Click here to download dataset (5.8Mb).
This dataset is identical to example in this repository.
-
Minimum test dataset
7 species, 100 CDS limited fasta dataset
FastDTLmapper -i example/minimum_dataset/fasta/ -t example/minimum_dataset/species_tree.nwk -o output_minimum -
Mycoplasma dataset (Input Format = Fasta)
7 Mycoplasma species, 500 ~ 1000 CDS fasta dataset
FastDTLmapper -i example/mycoplasma_dataset/fasta/ -t example/mycoplasma_dataset/species_tree.nwk -o output_mycoplasma_fasta -
Mycoplasma dataset (Input Format = Genbank)
7 Mycoplasma species, 500 ~ 1000 CDS genbank dataset
FastDTLmapper -i example/mycoplasma_dataset/genbank/ -t example/mycoplasma_dataset/species_tree.nwk -o output_mycoplasma_genbank
Output Contents
Output Top Directory
| Top directory | Contents | | ----------------------- | ------------------------------------------------------------ | | 00_user_data | Formatted user input fasta and tree files | | 01_orthofinder | OrthoFinder raw output results | | 02_dtl_reconciliation | Each OG(Ortholog Group) DTL reconciliation result | | 03_aggregate_map_result | Genome-wide DTL reconciliation aggregated and mapped results | | log | Config log and command log files |
Output Directory Structure & Files
.
├── 00_user_data/
Related Skills
node-connect
353.1kDiagnose OpenClaw node connection and pairing failures for Android, iOS, and macOS companion apps
claude-opus-4-5-migration
111.6kMigrate prompts and code from Claude Sonnet 4.0, Sonnet 4.5, or Opus 4.1 to Opus 4.5
frontend-design
111.6kCreate distinctive, production-grade frontend interfaces with high design quality. Use this skill when the user asks to build web components, pages, or applications. Generates creative, polished code that avoids generic AI aesthetics.
model-usage
353.1kUse CodexBar CLI local cost usage to summarize per-model usage for Codex or Claude, including the current (most recent) model or a full model breakdown. Trigger when asked for model-level usage/cost data from codexbar, or when you need a scriptable per-model summary from codexbar cost JSON.
