Igblast
Detailed explanation on how to setup database for NCBI IgBlast executable, and using it in Python
Install / Use
/learn @xinyu-dev/IgblastREADME
Using IgBlast with Python
Since IgBlast documentation does not have great details on the database setup, and there are a lot of questions on the forum regarding to the issue internal_data could not be found or the Germline annotation database...could not be found in [internal_data] directory, I wrote this short guide to in case it might be helpful.
This first part of this guide applies to installation of IgBlast and IMGT human germline sequence database. The second part applies to calling the IgBlast executable inside Jupyter notebook and converting some of the results to Pandas dataframe.
This repository contains executables and data files from ncbi-igblast-1.17.0-x64-macosx.tar.gz (latest Mac version as of Jan 9, 2021). It also has the IGHV, IGKV, and IGLV germline protein database obtained from IMGT (latest as of Jan 9, 2021).
Download IgBlast
Download executable here: ftp://ftp.ncbi.nih.gov/blast/executables/igblast/release/LATEST
Mac: Download the ncbi-igblast-1.17.0-x64-macosx.tar.gz or the latest version(NOT the dmg file) and unzip it in your destination folder.
Then:
- Create jupyter notebook inside the igblast folder.
- Create an empty folder called database inside the igblast folder
It should have a structure similar to what's shown below:
----igblast
--bin
--internal_data
--optional_file
--ChangeLog
--LICENSE
--ncbi_pakacage_info
--README
--database
--Using IgBlast.ipynb
Download IMGT germline sequence
- Germline seuqence can be found here. Both nucleic acid and protein sequences are available. If you
Note that the for blastp (protein blast), only V region alignment is provided by IgBlast.
For example, if you want to download the amino acid germline sequence for human IgHV, you can move to this section on the page, and click on Human under F+ORF+in-frame P. Note that that you can also select Human under F+ORF+in-frame P with IMGT gaps (sequence with gaps), but after cleaning the results will be the same.

- Once you click on the species in the page above, you will be directed to a page with a list of fasta sequences. Copy the fasta sequences.
For example, if we want to set up a database for human IGKV germline sequence, we would copy all fasta sequences by locating IGKV row and column F+ORF+in-frame P in table above, then click on the Human link. As of now, there is 108 sequences (example of the first 2 are shown below), and you need to copy all of them.
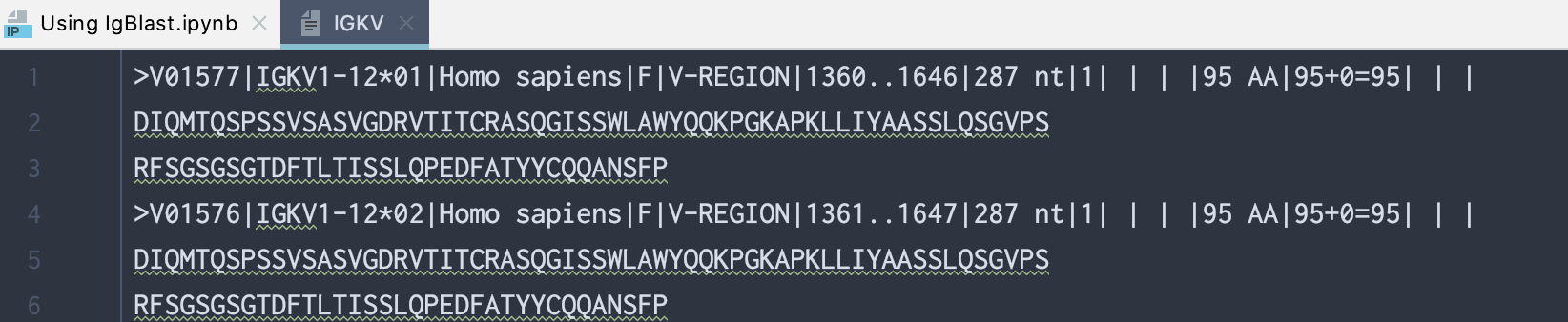
>V01577|IGKV1-12*01|Homo sapiens|F|V-REGION|1360..1646|287 nt|1| | | |95 AA|95+0=95| | |
DIQMTQSPSSVSASVGDRVTITCRASQGISSWLAWYQQKPGKAPKLLIYAASSLQSGVPS
RFSGSGSGTDFTLTISSLQPEDFATYYCQQANSFP
>V01576|IGKV1-12*02|Homo sapiens|F|V-REGION|1361..1647|287 nt|1| | | |95 AA|95+0=95| | |
DIQMTQSPSSVSASVGDRVTITCRASQGISSWLAWYQQKPGKAPKLLIYAASSLQSGVPS
......
......
- Inside the database folder you created in the 1st step, create another folder called Homo_sapiens and Homo_sapiens_clean Inside each of these 2 folders, create a folder called IG_dna and a folder called IG_prot. See screenshot below:
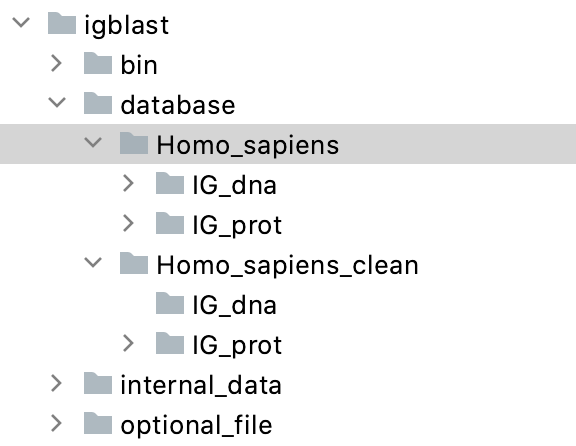
These folders are empty for now.
- Inside the IG_prot folder under Homo_sapiens folder, create a text file and paste the fasta sequences of IGKV in there.
Do NOT keep the .txt or .fasta extension for the text file. (otherwise Igblast automatically adds other extensions after your current extension, and your database name will look weird).
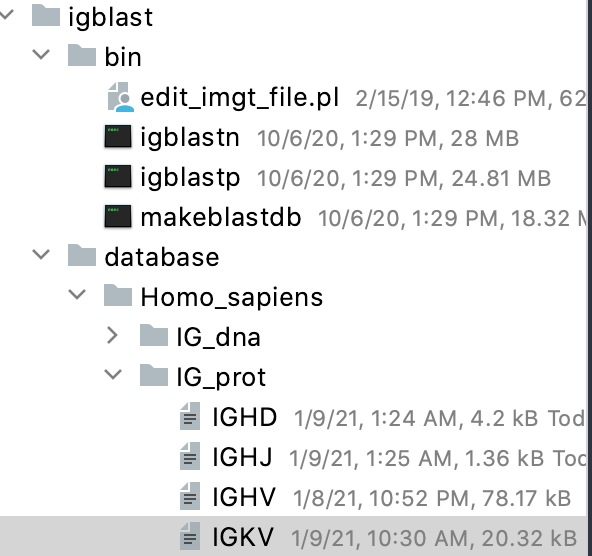
Here is what the directory structure looks like. Note that the text files do not have any extension (.txt or .fasta)
Here is a screenshot of what the IGKV text file looks like:
- If you have any DNA germline sequence, you can use similar method and copy them to IG_dna folder
Cleaning the IMGT germline sequence files
You can execute the command through terminal, or using the python script below.
Method 1: Use terminal
Open a terminal at the igblast folder. Then type
bin/edit_imgt_file.pl database/Homo_sapiens/IG_prot/IGKV > database/Homo_sapiens_clean/IG_prot/IGHV_clean
The above line cleans up the IGKV text file you just made, and save the cleaned file inside Homo_sapiens_clean/IG_prot folder.
After cleaning, the id, name, and description of the original IMGT fasta sequences are truncated to keep only the allele type. An example of cleaned sequence:

Method 2: Use python
# cleaning up IGKV IMGT germline sequence file
input_imgt_ref = 'database/Homo_sapiens/IG_prot/IGKV'
output_imgt_ref = 'database/Homo_sapiens_clean/IG_prot/IGKV_clean'
cmd = ['bin/edit_imgt_file.pl', input_imgt_ref , '>', output_imgt_ref]
# display result (and any error) in notebook. No file will be saved
# subprocess.run(cmd.split(), capture_output=True)
# save cmd output to file
fh = open(output_imgt_ref, 'w')
subprocess.Popen(cmd, stdout=fh)
Make Database
We need to parse the cleaned germline sequence file to database. There are 2 ways to do it. In terminal or in Python.
The basic syntax for DNA and protein seuqence is:
bin/makeblastdb -parse_seqids -dbtype nucl -in my_seq_file
bin/makeblastdb -parse_seqids -dbtype prot -in my_seq_file
Method 1: Use terminal
To parse the IGKV protein germline seuqences, open a terminal at the igblast folder. Then type
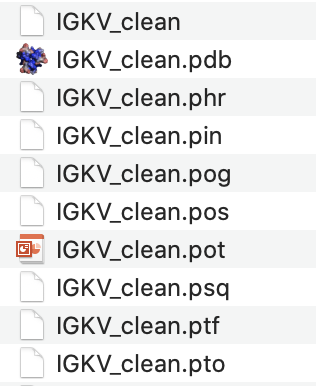
bin/makeblastdb -parse_seqids -dbtype prot -in database/Homo_sapiens_clean/IG_prot/IGKV_clean
Igblast will create a variety of files in the same directory as the cleaned germline sequence text file. A screenshot is shown below:
Method 2: Use Python
cmd = ['bin/makeblastdb', '-parse_seqids', '-dbtype', 'prot', '-in', output_imgt_ref]
subprocess.Popen(cmd, stdout=subprocess.PIPE)
Repeat the process for other germline sequence database. Create one database for VH, one for VL, one for VK...
Parse your sequence
Details of different parameters and options can be found using:
bin/iglbastp -help
bin/igblastn -help
Below is an example output of bin/igblastp -help
USAGE
igblastp [-h] [-help] [-germline_db_V germline_database_name]
[-num_alignments_V int_value] [-germline_db_V_seqidlist filename]
[-organism germline_origin] [-domain_system domain_system]
[-ig_seqtype sequence_type] [-focus_on_V_segment] [-extend_align5end]
[-extend_align3end] [-min_V_length Min_V_Length] [-db database_name]
[-dbsize num_letters] [-entrez_query entrez_query] [-query input_file]
[-out output_file] [-evalue evalue] [-word_size int_value]
[-gapopen open_penalty] [-gapextend extend_penalty] [-searchsp int_value]
[-sum_stats bool_value] [-matrix matrix_name] [-threshold float_value]
[-ungapped] [-lcase_masking] [-query_loc range] [-parse_deflines]
[-outfmt format] [-show_gis] [-num_descriptions int_value]
[-num_alignments int_value] [-line_length line_length]
[-num_threads int_value] [-remote] [-version]
DESCRIPTION
BLAST for Ig and TCR sequences
OPTIONAL ARGUMENTS
-h
Print USAGE and DESCRIPTION; ignore all other parameters
-help
Print USAGE, DESCRIPTION and ARGUMENTS; ignore all other parameters
-version
Print version number; ignore other arguments
*** Input query options
-query <File_In>
Input file name
Default = `-'
-query_loc <String>
Location on the query sequence in 1-based offsets (Format: start-stop)
*** General search options
-db <String>
Optional additional database name
-out <File_Out>
Output file name
Default = `-'
-evalue <Real>
Expectation value (E) threshold for saving hits
Default = `1'
-word_size <Integer, >=2>
Word size for wordfinder algorithm
-gapopen <Integer>
Cost to open a gap
-gapextend <Integer>
Cost to extend a gap
-matrix <String>
Scoring matrix name (normally BLOSUM62)
-threshold <Real, >=0>
Minimum word score such that the word is added to the BLAST lookup table
*** Formatting options
-outfmt <String>
alignment view options:
3 = Flat query-anchored, show identities,
4 = Flat query-anchored, no identities,
7 = Tabular with comment lines
19 = Rearrangement summary report (AIRR format)
Options 7 can be additionally configured to produce
a custom format specified by space delimited format specifiers.
The supported format specifiers are:
qseqid means Query Seq-id
qgi means Query GI
qacc means Query accesion
qaccver means Query accesion.version
qlen means Query sequence length
sseqid means Subject Seq-id
sallseqid means All subject Seq-id(s), separated by a ';'
sgi means Subject GI
sallgi means All subject GIs
sacc means Subject accession
saccver means Subject accession.version
sallacc means All subject accessions
slen means Subject sequence length
qstart means Start of alignment in query
qend means End of alignment in query
sstart means Start of alignment in subject
send means End of alignment in subject
qseq means Align
