Ramanbiolib
A Raman spectral search library for biological molecules identification, over a database of 140 components, including lipids, proteins, carbohydrates, amino acids, metabolites, nucleic acids, pigments and others.
Install / Use
/learn @mteranm/RamanbiolibREADME
RamanBiolib
A Python-based Raman spectral search library for biological molecules identification, over a database of 140 components, including lipids, proteins, carbohydrates, amino acids, metabolites, nucleic acids, pigments and others.
The library supports identification via spectral similarity or peak-matching algorithms, using either full spectrum plots or most relevant peak positions. For details on the underlying methods, see our article.
Installation
pip install ramanbiolib
RamanBiolib Graphical User Interface (GUI)
For the GUI see the project RamanBiolib-UI
Usage
Spectra similarity search
Identify an unkown component by its Raman spectrum.
spectra_search = SpectraSimilaritySearch(wavenumbers=wavenumbers)
search_results = spectra_search.search(
unknown_spectrum,
class_filter=None,
unique_components_in_results=True,
similarity_method="slk",
similarity_params=25 # Window size
)
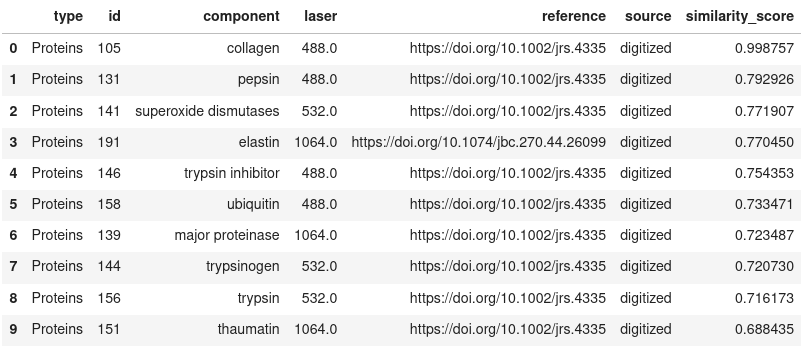
Show the top N results
search_results.get_results(limit=10)
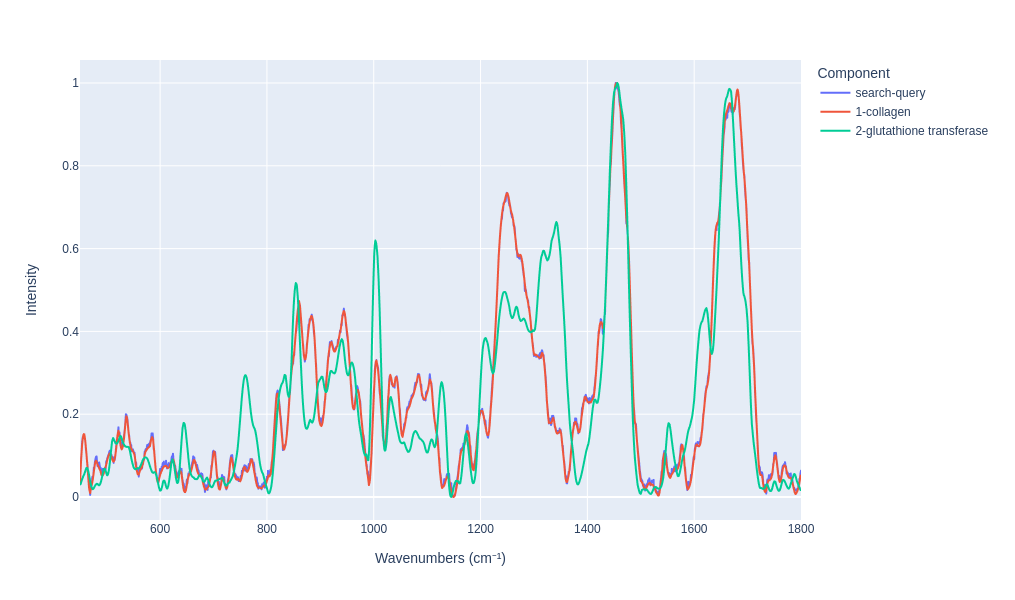
Visualy compare the results
search_results.plot_results(n=2)
Peak matching search
Identify an unkown component by its Raman spectrum most relevant peaks positions.
pm_search = PeakMatchingSearch(wavenumbers=wavenumbers)
pm_search_results = pm_search.search(
peaks_wavenumbers,
tolerance=5,
class_filter=None,
sort_score='IUR',
min_peak_intensity=0,
tol_penalty="linear",
unique_components_in_results=True
)
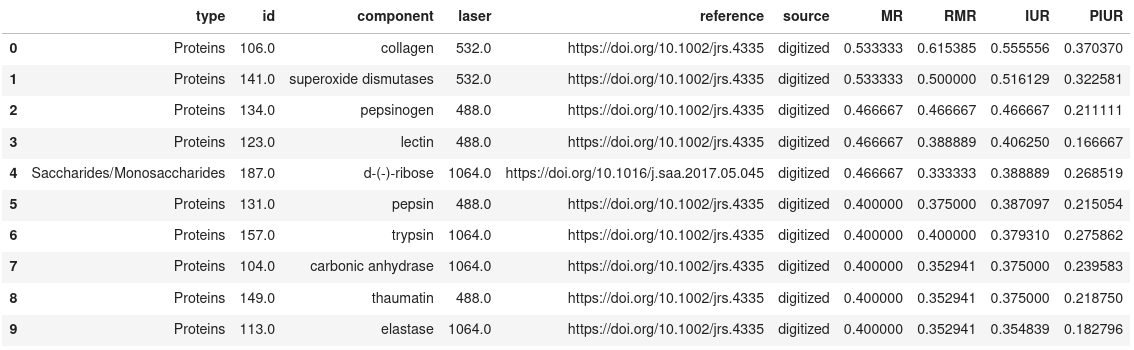
Show the top N results
pm_search_results.get_results(limit=10, sort_col='IUR')
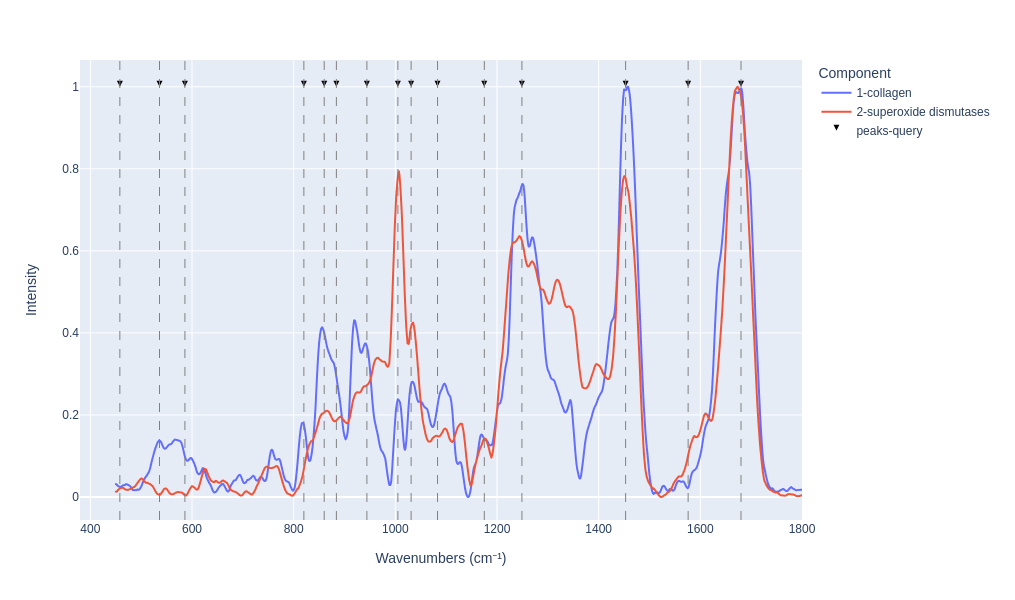
Visualy compare the results
pm_search_results.plot_results(n=2)
Examples
Jupyter notebooks search example:
API documentation
How to contribute with Raman spectra
Note: All contributed spectral data will be licensed under ODbL open database license. See the license section for details.
The collaborative effort of the research community is essential to expanding this open spectral library for biomolecule identification. We welcome your contributions through the following methods:
Measured spectra (✅ Preferred Option)
Spectra measured in the laboratory can be included if they are from an isolated pure sample of a biomolecule.
Articles plots digitazion
As done in the initial version of this database, you can contribute to the digitazion process of the already published Raman spectra in articles figures.
Important: Only spectra digitized from figures in published articles are accepted.
Contribution process
- Fork this repository.
- Create a branch (feature/branch_name)
- Add your new spectra entries to:
ramanbiolib/db/raman_spectra_db.csvramanbiolib/db/raman_peaks_db.csvramanbiolib/db/metadata_db.csv
- Commit & Push your changes.
- Open a Pull Request (PR).
- Once reviewed and approved, your contribution will be included in the next database release.
PR useful docs:
To contribute, you need to add new rows with the new spectra information in all database files: raman_spectra_db.csv, raman_peaks_db.csv, and metadata_db.csv.
Mandatory Fields: (M) are mandatory fields, (R) are optional but recommended.
Spectra plot (raman_spectra_db.csv)
- id (M): the identifier of the spectrum in this database. This is a incremental id, therefore new specturm will be identified with max_current_id + 1. (This id needs to match with the other files id for the same spectrum measurement)
- component (M): component name (in lower case, if the component is already existing in the database use the same name)
- wavenumbers (M): a list of the spectrum wavenumbers, minimum from 450 to 1800 with a step of 1cm⁻¹ (e.g., "[450, 451, 452, 453, 454, 455, ... , 1800]")
- intensity (M): a list of the spectrum intensity values (e.g., "[0.021540, 0.0215406, ..., 0.0137668])
The spectra needs to be standarized as follows:
Standarization:
- Smoothed to reduce noise
- Baseline removed
- Interpolated to minimum 450-1800 cm⁻¹ with a step of 1 cm⁻¹
- Min-max normalized
Peak positions list (raman_peaks_db.csv)
- id (M): the identifier of the spectrum in this database. This is a incremental id, therefore new specturm will be identified with max_current_id + 1. (This id need to match with the other files id for the same spectrum measurement)
- component (M): component name (in lower case, if the component is already existing in the database use the same name)
- peaks (M): the position (in wavenumbers cm⁻¹) of the most relevant peaks, integer values (e.g., "[536, 560, 623, ..., 1612]"). All relevant peaks within the range 450 to 1800 cm⁻¹ need to be added.
- intensity (M): the list of the intensity of the most relevan peaks, after min-max normalization, the list size and order need to match the previous column size. (e.g., "[0.021540, 0.021540, ..., 0.013766])
Metadata (metadata_db.csv)
- id (M): the identifier of the spectrum in this database. This is a incremental id, therefore new specturm will be identified with max_current_id + 1. (This id need to match with the other database id for the same spectrum measurement)
- component (M): component name (in lower case, if the component is already existing in the database use the same name)
- type (M): the molecule type tree separated by slash '/', pelase check current types to keep the same structure (e.g., Lipids/FattyAcids)
- submission_date (M): the submission date in format YYYY-MM-DD,
- contact (M): contact data of the person/team who submits data,
- source (M): the value 'digitized' if you obatined the spectra from a digitazion process from article figures, the value 'measured' if are lab measurements.
- reference (M): the reference DOI to the article associated with the data. If the spectrum was digitized, it indicates the article from which the spectrum was obtained. In the case of measurements, this field is mandatory only if there is a related publication.
- extraction_method (M): if spectra were obtained from an external source, specify how it was extracted (digitalization method).
- peak_identificaton (M): method implemented to detect the Raman bands or peaks.,
- interpolation_method (M): interpolation method used to match the wavenumbers of the database.
- extra_preprocessing (M): the specification of additional data treatment implemented after extracting spectra from the external reference and before submitting.
- complete_sample_name (M): the complete name of the sample.
- sample_source (M): the origin of the sample, such as, commercial supplier, commercial reference, environment or tissue provenance.
- sample_composition (M): the sample composition when measured, such as medium, purity and concentration.
- sample_preparation (R): the sample processing performed on the original sample. Also, conditions in which the sample is measured, such as temperature, pH or humidity, etc.
- sample_substrate (M): the components that were used to measure the sample that may introduce further contributions to spectra. For instance, glass, quartz, CaF2 coverslips, microscope slides or cuvettes. In the case of implementing SERS, the nanoparticles and/or the substrate implemented must be specified.
- raman_technique (M): the Raman tecnique used. For instance, spontaneous Raman spectroscopy, Fourier Transform Raman spectroscopy, Surface Enhanced Raman spectroscopy (SERS), Tip Enhanced Raman spectroscopy (TERS), Spatially-Offset Raman spectroscopy (SORS), time-gated Raman, etc.
- raman_system (M): If commercial, provide manufacturer and model reference. If custom built, specify it (M), and provide the commercial references of the different optical components (R). Provide the details, if possible, of laser, monochromator and detector models.
- delivery_optics (M): the delivery/collection optics. If an objective was used, provide commercial reference, numerical aperture, magnification, immersion type. If a fiber-optic Raman probe is used, provide commercial reference, core diameters, numerical aperture if applied, spot size, and probe geometry. Also specify if a pinhole was used for confocality
- **laser_wav
Related Skills
feishu-drive
332.3k|
things-mac
332.3kManage Things 3 via the `things` CLI on macOS (add/update projects+todos via URL scheme; read/search/list from the local Things database)
clawhub
332.3kUse the ClawHub CLI to search, install, update, and publish agent skills from clawhub.com
convex_rules
--- description: Guidelines and best practices for building Convex projects, including database schema design, queries, mutations, and real-world examples globs: / .ts, / .tsx, / .js, / .jsx -
