PyGenomeViz
A genome visualization python package for comparative genomics
Install / Use
/learn @moshi4/PyGenomeVizREADME
pyGenomeViz


![]()
[!NOTE] A major version upgrade, pyGenomeViz v1.0.0, was released on 2024/05. Backward incompatible changes have been made between v1.0.0 and v0.X.X to make for a more sophisticated API/CLI design. Therefore, v0.X.X users should pin the version to v0.4.4 or update existing code for v1.0.0. Previous v0.4.4 documentation is available here.
Table of contents
- Overview
- Installation
- API Examples
- CLI Examples
- GUI (Web Application)
- HTML Viewer
- Inspiration
- Circular Genome Visualization
- Star History
Overview
pyGenomeViz is a genome visualization python package for comparative genomics implemented based on matplotlib. This package is developed for the purpose of easily and beautifully plotting genomic features and sequence similarity comparison links between multiple genomes. It supports genome visualization of Genbank/GFF format file and can be saved figure in various formats (JPG/PNG/SVG/PDF/HTML). User can use pyGenomeViz for interactive genome visualization figure plotting on jupyter notebook, or automatic genome visualization figure plotting in genome analysis scripts/workflow.
For more information, please see full documentation here.
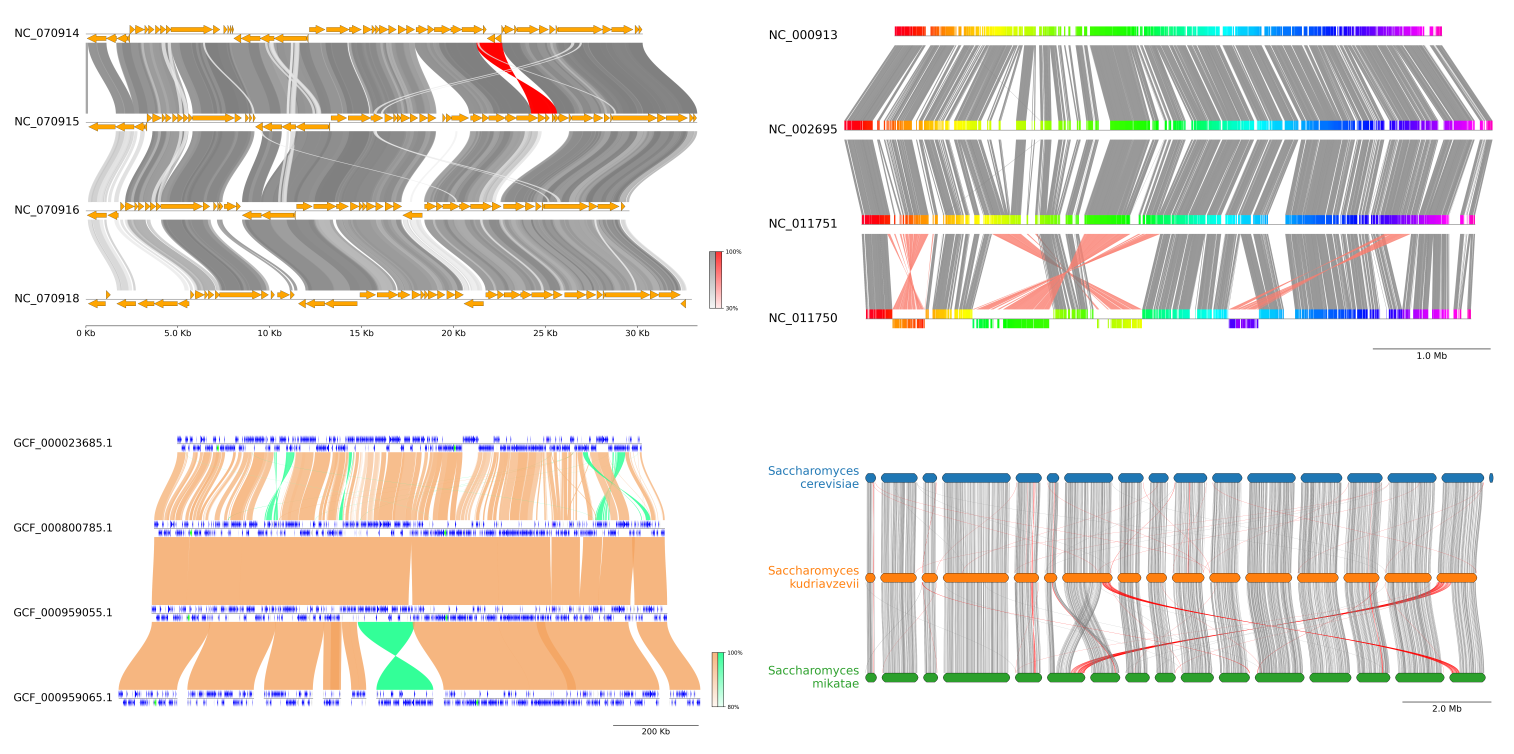
Fig.1 pyGenomeViz example plot gallery
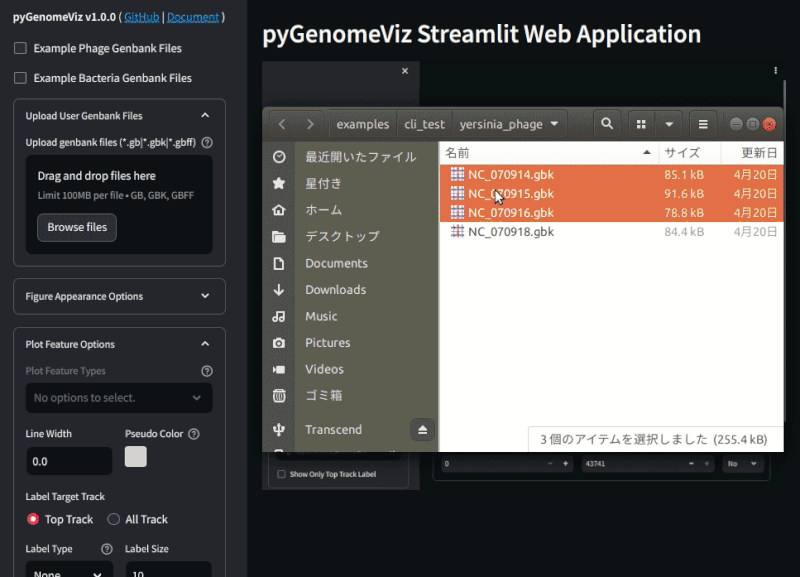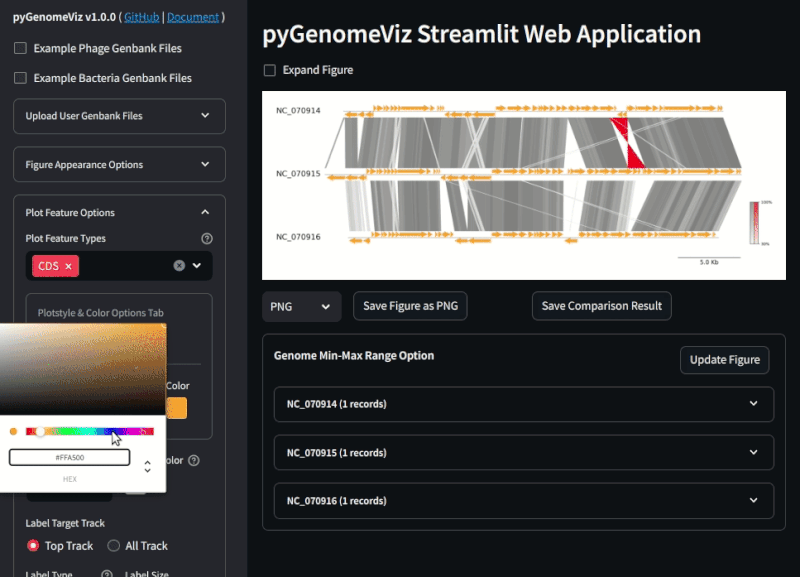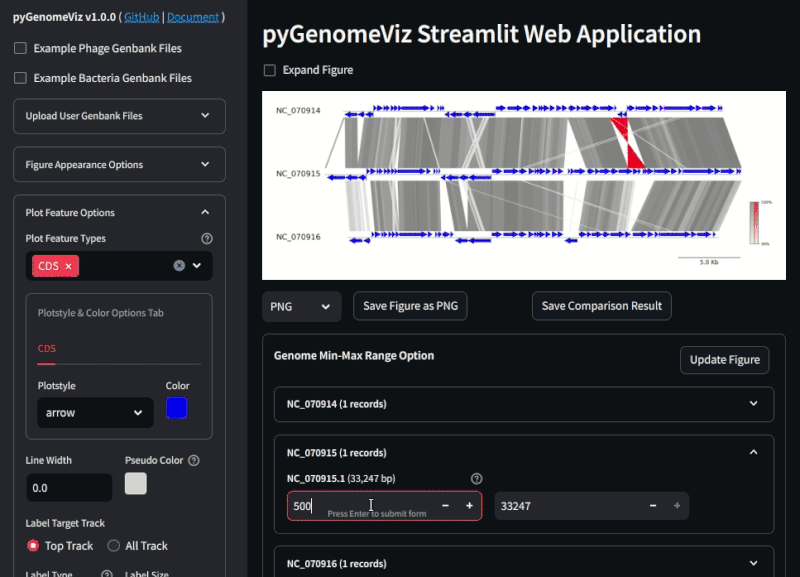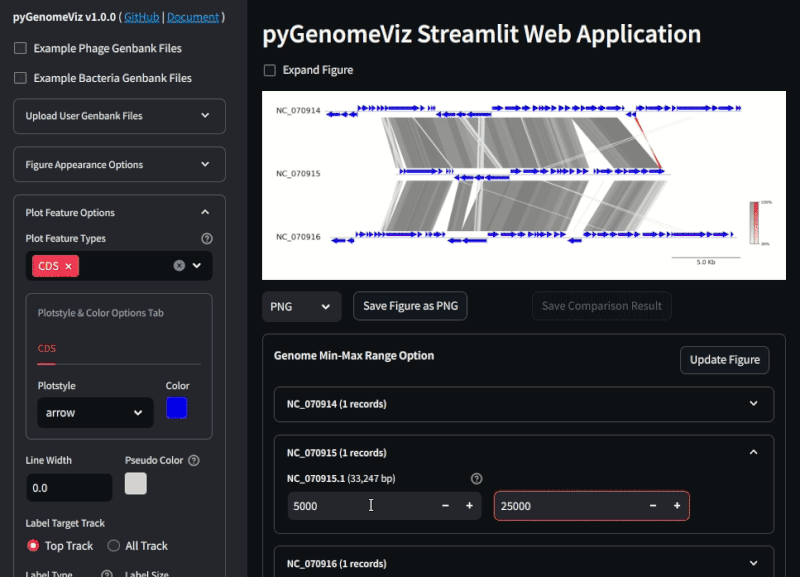 Fig.2 pyGenomeViz web application example (Demo Page)
Fig.2 pyGenomeViz web application example (Demo Page)
Installation
Python 3.9 or later is required for installation.
Install PyPI package:
pip install pygenomeviz
Install conda-forge package:
conda install -c conda-forge pygenomeviz
Use Docker (Image Registry):
docker run -it --rm -p 8501:8501 ghcr.io/moshi4/pygenomeviz:latest pgv-gui -h
API Examples
Jupyter notebooks containing code examples below is available here.
Features
from pygenomeviz import GenomeViz
gv = GenomeViz()
gv.set_scale_xticks(ymargin=0.5)
track = gv.add_feature_track("tutorial", 1000)
track.add_sublabel()
track.add_feature(50, 200, 1)
track.add_feature(250, 460, -1, fc="blue")
track.add_feature(500, 710, 1, fc="lime")
track.add_feature(750, 960, 1, fc="magenta", lw=1.0)
gv.savefig("features.png")
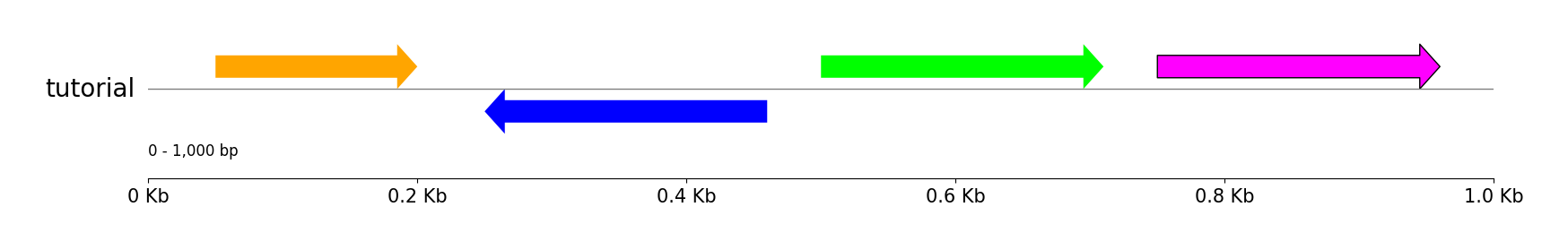
Styled Features
from pygenomeviz import GenomeViz
gv = GenomeViz()
gv.set_scale_bar(ymargin=0.5)
track = gv.add_feature_track("tutorial", (1000, 2000))
track.add_sublabel()
track.add_feature(1050, 1150, 1, label="arrow")
track.add_feature(1200, 1300, -1, plotstyle="bigarrow", label="bigarrow", fc="red", lw=1)
track.add_feature(1330, 1400, 1, plotstyle="bigbox", label="bigbox", fc="blue", text_kws=dict(rotation=0, hpos="center"))
track.add_feature(1420, 1500, 1, plotstyle="box", label="box", fc="limegreen", text_kws=dict(size=10, color="blue"))
track.add_feature(1550, 1600, 1, plotstyle="bigrbox", label="bigrbox", fc="magenta", ec="blue", lw=1, text_kws=dict(rotation=0, vpos="bottom", hpos="center"))
track.add_feature(1650, 1750, -1, plotstyle="rbox", label="rbox", fc="grey", text_kws=dict(rotation=-45, vpos="bottom"))
track.add_feature(1780, 1880, 1, fc="lime", hatch="o", arrow_shaft_ratio=0.2, label="arrow shaft\n0.2", text_kws=dict(rotation=0, hpos="center"))
track.add_feature(1890, 1990, 1, fc="lime", hatch="/", arrow_shaft_ratio=1.0, label="arrow shaft\n1.0", text_kws=dict(rotation=0, hpos="center"))
gv.savefig("styled_features.png")
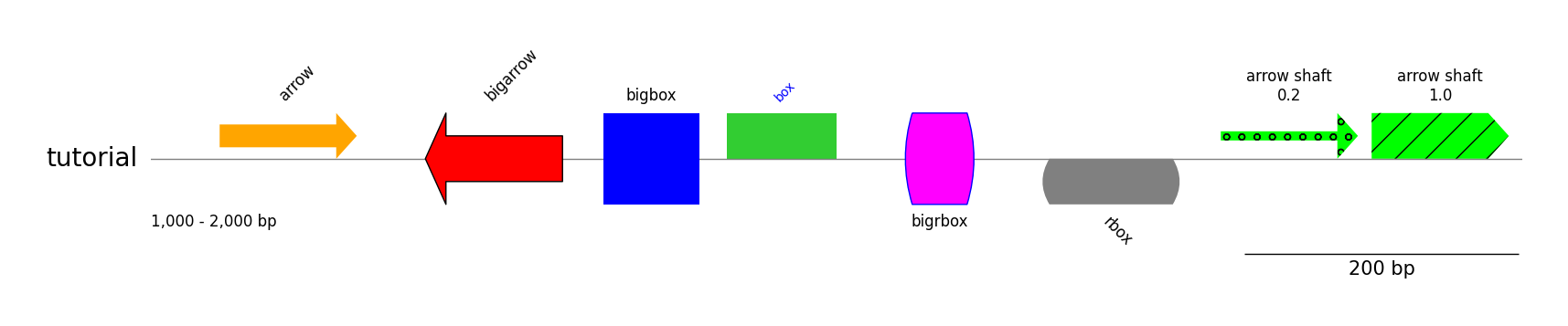
Tracks & Links
from pygenomeviz import GenomeViz
genome_list = [
dict(name="genome 01", size=1000, features=((150, 300, 1), (500, 700, -1), (750, 950, 1))),
dict(name="genome 02", size=1300, features=((50, 200, 1), (350, 450, 1), (700, 900, -1), (950, 1150, -1))),
dict(name="genome 03", size=1200, features=((150, 300, 1), (350, 450, -1), (500, 700, -1), (700, 900, -1))),
]
gv = GenomeViz(track_align_type="center")
gv.set_scale_bar()
for genome in genome_list:
name, size, features = genome["name"], genome["size"], genome["features"]
track = gv.add_feature_track(name, size)
track.add_sublabel()
for idx, feature in enumerate(features, 1):
start, end, strand = feature
track.add_feature(start, end, strand, plotstyle="bigarrow", lw=1, label=f"gene{idx:02d}", text_kws=dict(rotation=0, vpos="top", hpos="center"))
# Add links between "genome 01" and "genome 02"
gv.add_link(("genome 01", 150, 300), ("genome 02", 50, 200))
gv.add_link(("genome 01", 700, 500), ("genome 02", 900, 700))
gv.add_link(("genome 01", 750, 950), ("genome 02", 1150, 950))
# Add links between "genome 02" and "genome 03"
gv.add_link(("genome 02", 50, 200), ("genome 03", 150, 300), color="skyblue", inverted_color="lime", curve=True)
gv.add_link(("genome 02", 350, 450), ("genome 03", 450, 350), color="skyblue", inverted_color="lime", curve=True)
gv.add_link(("genome 02", 900, 700), ("genome 03", 700, 500), color="skyblue", inverted_color="lime", curve=True)
gv.add_link(("genome 03", 900, 700), ("genome 02", 1150, 950), color="skyblue", inverted_color="lime", curve=True)
gv.savefig("tracks_and_links.png")
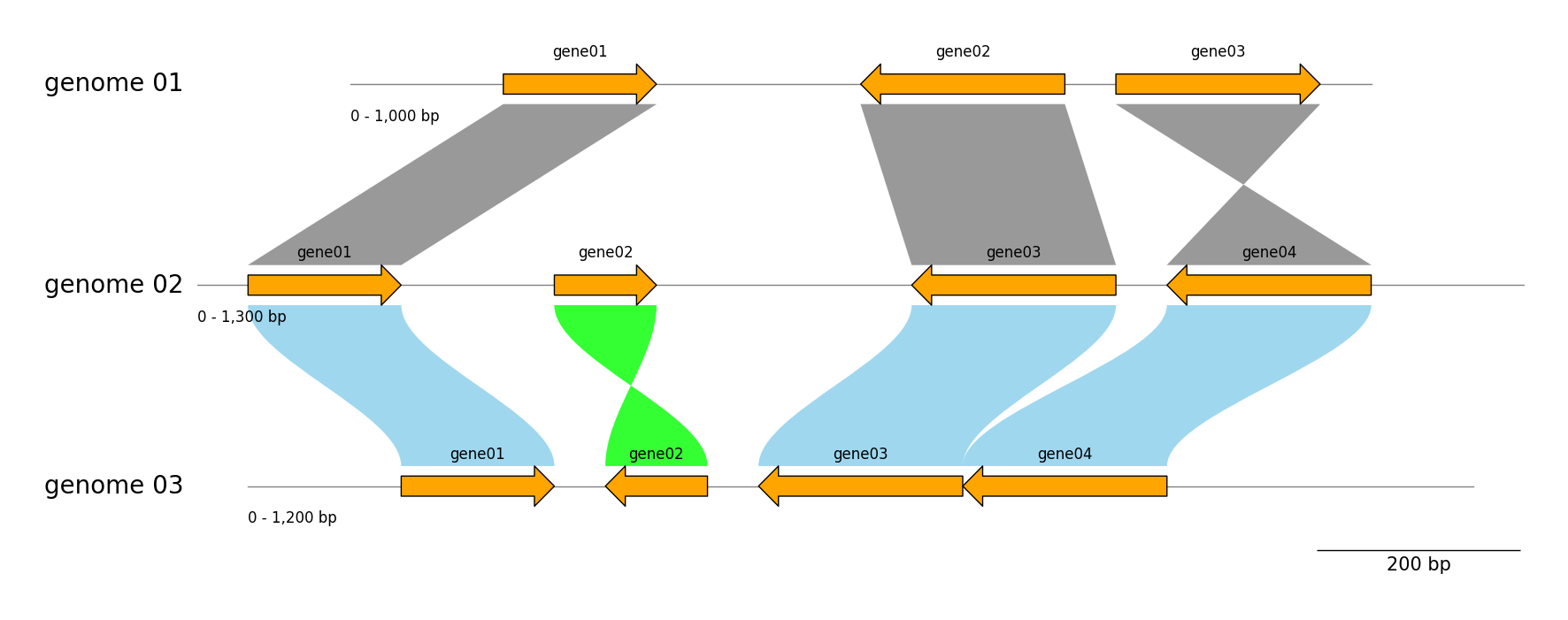
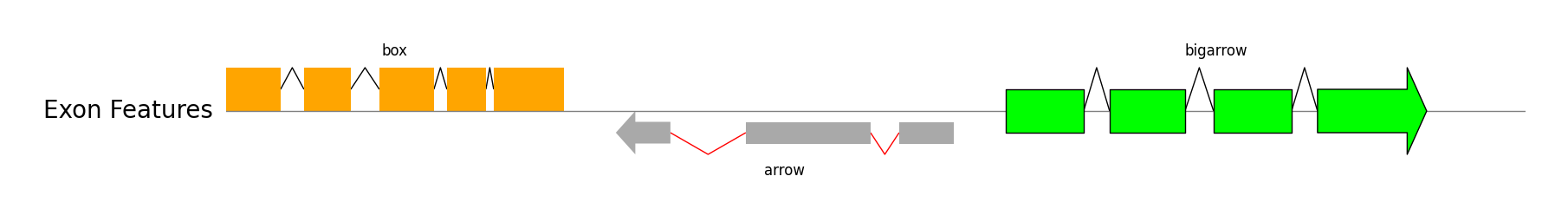
Exon Features
from pygenomeviz import GenomeViz
exon_regions1 = [(0, 210), (300, 480), (590, 800), (850, 1000), (1030, 1300)]
exon_regions2 = [(1500, 1710), (2000, 2480), (2590, 2800)]
exon_regions3 = [(3000, 3300), (3400, 3690), (3800, 4100), (4200, 4620)]
gv = GenomeViz()
track = gv.add_feature_track("Exon Features", 5000)
track.add_exon_feature(exon_regions1, strand=1, plotstyle="box", label="box", text_kws=dict(rotation=0, hpos="center"))
track.add_exon_feature(exon_regions2, strand=-1, plotstyle="arrow", label="arrow", text_kws=dict(rotation=0, vpos="bottom", hpos="center"), patch_kws=dict(fc="darkgrey"), intron_patch_kws=dict(ec="red"))
track.add_exon_feature(exon_regions3, strand=1, plotstyle="bigarrow", label="bigarrow", text_kws=dict(rotation=0, hpos="center"), patch_kws=dict(fc="lime", lw=1))
gv.savefig("exon_features.png")
Genbank Features
from pygenomeviz import GenomeViz
from pygenomeviz.parser import Genbank
from pygenomeviz.utils import load_example_genbank_dataset
gbk_files = load_example_genbank_dataset("yersinia_phage")
gbk = Genbank(gbk_files[0])
gv = GenomeViz()
gv.set_scale_bar(ymargin=0.5)
track = gv.add_feature_track(gbk.name, gbk.genome_length)
track.add_sublabel()
features = gbk.extract_features()
track.add_features(features)
gv.savefig("genbank_features.png")
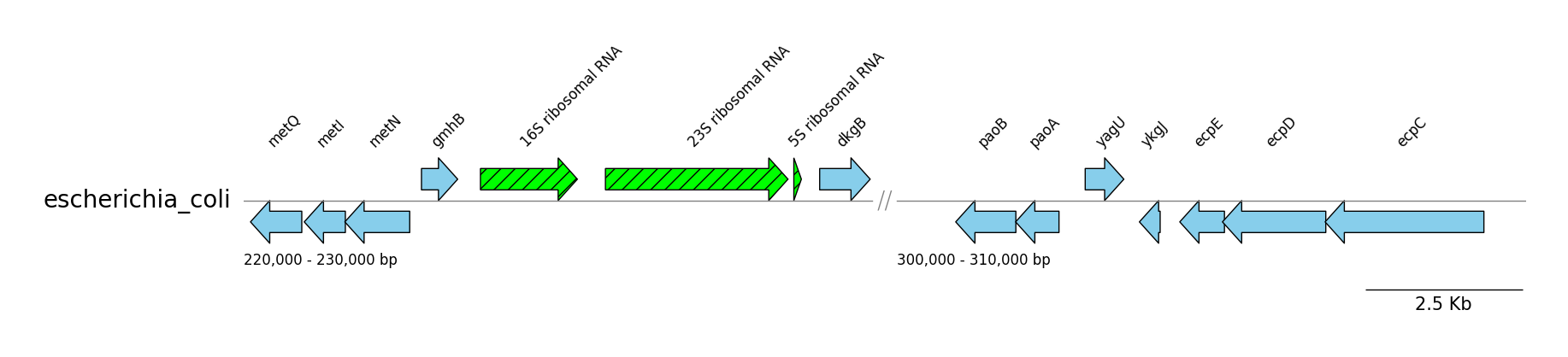
GFF Features
from pygenomeviz import GenomeViz
from pygenomeviz.parser import Gff
from pygenomeviz.utils import load_example_gff_file
gff_file = load_example_gff_file("escherichia_coli.gff.gz")
gff = Gff(gff_file)
gv = GenomeViz()
gv.set_scale_bar(ymargin=0.5)
target_ranges = ((220000, 230000), (300000, 310000))
track = gv.add_feature_track(name=gff.name, segments=target_ranges)
track.set_segment_sep(symbol="//")
for segment in track.segments:
segment.add_sublabel()
# Plot CDS features
cds_features = gff.extract_features(feature_type="CDS", target_range=segment.range)
segment.add_features(cds_features, label_type="gene", fc="skyblue", lw=1.0)
# Plot rRNA features
rrna_features = gff.extract_features(feature_type="rRNA", target_range=segment.range)
segment.add_features(rrna_features, label_type="product", hatch="//", fc="lime", lw=1.0)
gv.savefig("gff_features.png")
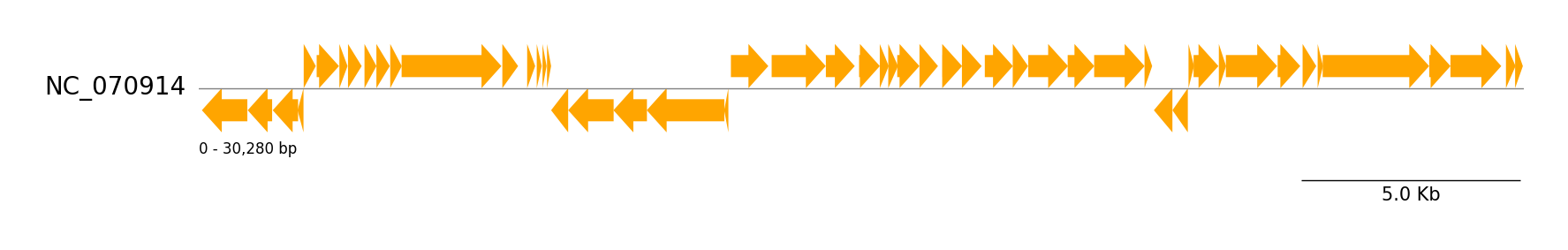
GFF Contigs
from pygenomeviz import GenomeViz
from pygenomeviz.parser import Gff
from pygenomeviz.utils import load_example_gff_file, is_pseudo_feature
gff_file = load_example_gff_file("mycoplasma_mycoides.gff")
gff = Gff(gff_file)
gv = GenomeViz(fig_track_height=0.5, feature_track_ratio=0.5)
gv.set_scale_xticks(labelsize=10)
# Plot CDS, rRNA features for each contig to tracks
for seqid, size in gff.get_seqid2size().items():
track = gv.add_feature_track(seqid, size, labelsize=15)
track.add_sublabel(size=10, color="grey")
cds_features = gff.get_seqid2features(feature_type="CDS")[seqid]
# CDS: blue, CDS(pseudo): grey
for cds_feature in cds_features:
color = "grey" if is_pseudo_feature(cds_feature) else "blue"
Related Skills
node-connect
345.9kDiagnose OpenClaw node connection and pairing failures for Android, iOS, and macOS companion apps
claude-opus-4-5-migration
106.4kMigrate prompts and code from Claude Sonnet 4.0, Sonnet 4.5, or Opus 4.1 to Opus 4.5
frontend-design
106.4kCreate distinctive, production-grade frontend interfaces with high design quality. Use this skill when the user asks to build web components, pages, or applications. Generates creative, polished code that avoids generic AI aesthetics.
model-usage
345.9kUse CodexBar CLI local cost usage to summarize per-model usage for Codex or Claude, including the current (most recent) model or a full model breakdown. Trigger when asked for model-level usage/cost data from codexbar, or when you need a scriptable per-model summary from codexbar cost JSON.
