Mmseq
Haplotype, isoform and gene level expression analysis using multi-mapping RNA-seq reads
Install / Use
/learn @eturro/MmseqREADME
MMSEQ: Transcript and gene level expression analysis using multi-mapping RNA-seq reads

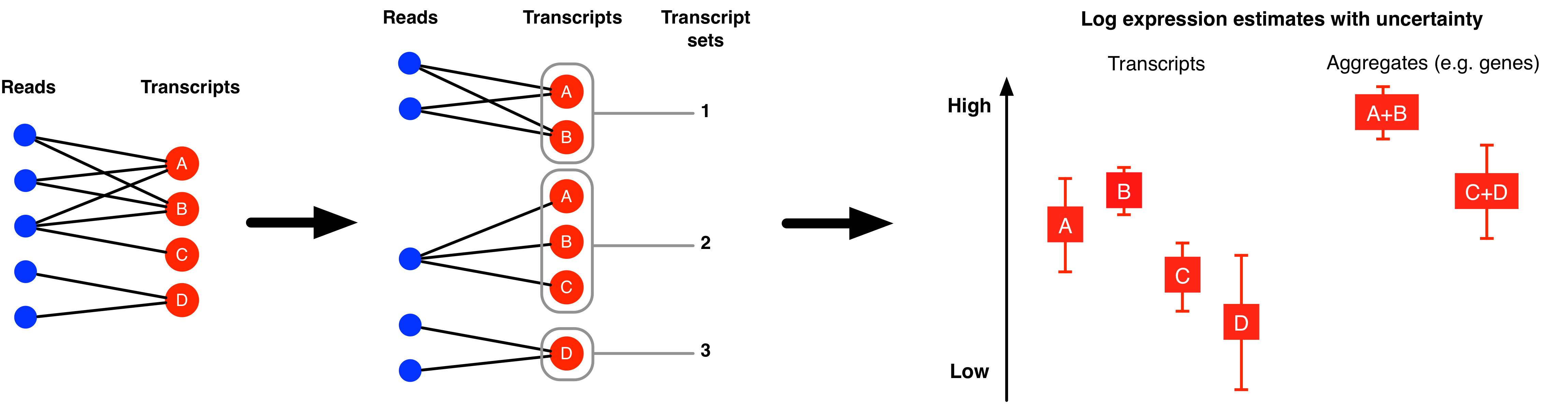
What is MMSEQ?
The MMSEQ package contains a collection of statistical tools for analysing RNA-seq expression data. Expression levels are inferred for each transcript using the mmseq program by modelling mappings of reads or read pairs (fragments) to sets of transcripts. These transcripts can be based on reference, custom or haplotype-specific sequences. The latter allows haplotype-specific analysis, which is useful in studies of allelic imbalance. The posterior distributions of the expression parameters for groups of transcripts belonging to the same gene are aggregated to provide gene-level expression estimates. Other aggregations (e.g. of transcripts sharing the same UTRs) are also possible. Isoform usage (i.e., the proportion of a gene's expression due to each isoform) is also estimated. Uncertainty in expression levels is summarised as the standard deviation of the posterior distribution of each expression parameter. When the uncertainty is large in all samples, a collapsing algorithm can be used for grouping transcripts into inferential units with reduced levels of uncertainty.
The package also includes a model-selection algorithm for differential analysis (implemented in mmdiff) that takes into account the posterior uncertainty in the expression parameters and can be used to select amongst an arbitrary number of models. The algorithm is regression-based and thus it can accomodate complex experimental designs. The model selection algorithm can be applied at the level of transcripts or transcript aggregates such as genes and it can also be applied to detect differential isoform usage by modelling summaries of the posterior distributions of isoform usage proportions as the outcomes of the linear regression models.
Citing MMSEQ
If you use the MMSEQ package, please cite:
- Haplotype and isoform specific expression estimation using multi-mapping RNA-seq reads. Turro E, Su S-Y, Goncalves A, Coin L, Richardson S and Lewin A. Genome Biology, 2011 Feb; 12:R13. doi: 10.1186/gb-2011-12-2-r13.
If you use the mmdiff or mmcollapse programs, please also cite:
- Flexible analysis of RNA-seq data using mixed effects models. Turro E, Astle WJ and Tavaré S. Bioinformatics, 2013. doi: 10.1093/bioinformatics/btt624.
Key features
- Isoform-level expression analysis (works out of the box with Ensembl cDNA and ncRNA files)
- Gene-level expression analysis that is robust to changes in isoform usage, unlike count-based methods
- Haplotype-specific analysis, useful for eQTL analysis and studies of F1 crosses
- Multi-mapping of reads, including mapping to transcripts from different genes, is properly taken into account
- The insert size distribution is taken into account
- Sequence-specific biases can be taken into account
- Flexible differential analysis based on linear mixed models
- Uncertainty in expression parameters is taken into account
- Polytomous model selection (i.e. selecting amongst numerous competing models)
- Modelling of isoform usage proportions
- Collapsing of transcripts with high levels of uncertainty into inferential units which can be estimated with reduced uncertainty
- Multi-threaded C++ implementations
Installation
Download the latest release of MMSEQ, unzip and add the bin directory to your PATH. E.g.:
wget -O mmseq-latest.zip https://github.com/eturro/mmseq/archive/latest.zip
unzip mmseq-latest.zip && cd mmseq-latest
export PATH=`pwd`/bin:$PATH
You might want to strip the suffix from the binaries. E.g., under Linux:
cd bin
for f in `ls *-linux`; do
mv $f `basename $f -linux`
done
The current release is 1.0.10 (changelog). Visit the release archive to download older releases.
Estimating expression levels
Input files:
- FASTQ files containing reads from the experiment
- A FASTA file containing transcript sequences to align to (find ready-made files)
The example commands below assume that the FASTQ files are asample_1.fq and asample_2.fq (paired-end) and the FASTA file is Homo_sapiens.GRCh37.70.ref_transcripts.fa.
Step 1a: Index the reference transcript sequences with Bowtie 1 (not Bowtie 2); to use kallisto instead, see Step 1b
bowtie-build --offrate 3 Homo_sapiens.GRCh37.70.ref_transcripts.fa Homo_sapiens.GRCh37.70.ref_transcripts
(It is advisable to use a lower-than-default value for --offrate (such as 2 or 3) as long as the resulting index fits in memory.)
Step 1b: Index the reference transcript sequences with kallisto (this is an alternative to Step 1a)
kallisto index -i Homo_sapiens.GRCh37.70.ref_transcripts.kind Homo_sapiens.GRCh37.70.ref_transcripts.fa
Step 1c (optional): Trim out adapter sequences if necessary
If the insert size distribution overlaps the read length, trim back the reads to exclude adapter sequences. Trim Galore! works well. E.g. for libraries prepared using standard Illumina adapters (AGATCGGAAGAGC), run:
trim_galore -q 15 --stringency 3 -e 0.05 --length 36 --trim1 --paired asample_1.fq.gz asample_2.fq.gz
Step 2a: Align reads with Bowtie 1 (not Bowtie 2); to use kallisto instead, see Step 2b
bowtie -a --best --strata -S -m 100 -X 500 --chunkmbs 256 -p 8 Homo_sapiens.GRCh37.70.ref_transcripts \
-1 <(gzip -dc asample_1.fq.gz) -2 <(gzip -dc asample_2.fq.gz) | samtools view -F 0xC -bS - | \
samtools sort -n - asample.namesorted
- Always specify
-ato ensure you get multi-mapping alignments - Suppress alignments for reads that map to a huge number of transcripts with the
-moption (e.g.-m 100) - Adjust
-Xaccording to the maximum insert size - Specify
--norcif the data were generated following a forward-stranded protocol - If the reference FASTA file doesn't use the Ensembl convention, then also specify
--fullref - The read names must end with /1 or /2, not /3 or /4 (this can be corrected with
awk 'FNR % 4==1 { sub(/\/[34]$/, "/2") } { print }' secondreads.fq > secondreads-new.fq). - If there are multiple FASTQ files from the same library, feed them all together to Bowtie in one go (delimit the FASTQ file names with commas)
- The command above assumes the FASTQ files are gzipped, hence the
gzip -dc - If you are getting many "Exhausted best-first chunk memory" warnings, try increasing
--chunkmbsto 128 or 256. - If the read names contain spaces, make sure the substring up to the first space in each read is unique, as Bowtie strips anything after a space in a read name
- The output BAM file must be sorted by read name.
- With paired-end data, only pairs where both reads have been aligned are used, so might as well use the samtools
0xCfiltering flag as above to reduce the size of the BAM file
Step 2b: Align reads with kallisto (this is an alternative to Step 2a)
kallisto pseudo -i Homo_sapiens.GRCh37.70.ref_transcripts.kind --pseudobam -o kout asample_1.fq.gz asample_2.fq.gz | \
| samtools view -F 0xC -bS - | samtools sort -n - asample.namesorted
Step 3: Map reads to transcript sets
bam2hits Homo_sapiens.GRCh37.70.ref_transcripts.fa asample.namesorted.bam > asample.hits
Step 4: Obtain expression estimates
mmseq asample.hits asample
Description of output:
-
asample.mmseqcontains a table with columns:- feature_id: name of transcript
- log_mu: posterior mean of the log_e expression parameter (use this as your log expression measure)
- sd: posterior standard deviation of the log_e expression parameter
- mcse: Monte Carlo standard error
- iact: integrated autocorrelation time
- effective_length: effective transcript length
- true_length: length of transcript sequence
- unique_hits: number of reads uniquely mapping to the transcript
- mean_proportion: posterior mean isoform/gene proportion
- mean_probit_proportion: posterior mean of the probit-transformed isoform/gene proportion
- sd_probit_proportion: posterior standard deviation of the probit-transformed isoform/gene proportion
- log_mu_em: log-scale transcript-level EM estimate
- observed: whether or not a feature has hits
- ntranscripts: number of isoforms for the gene of that transcript
-
asample.identical.mmseq: as above but aggregated over transcripts sharing the same sequence (these estimates are usually far more precise than the corresponding individual estimates in the transcript-level table); note thatlog_mu_emand proportion summaries are not available for aggregates -
asample.gene.mmseq: as above but aggregated over genes and theeffective_lengthis an average of isoform effective lengths weighted by their expression -
Various other files (
asample.*.trace_gibbs.gz,asample.Mandasample.k) containing more detailed output
These steps operate on a sample-by-sample basis and the expression estimates are roughly proportional to the RNA concentrations in each sample. Some scaling of the estimates may be required to make them comparable across biological replicates and conditions. This can be achieved by reading in the output for multiple samples using the readmmseq() function defined in the mmseq.R R script included in the src/R directory (more information on this function can be found [here](ht
Related Skills
node-connect
341.2kDiagnose OpenClaw node connection and pairing failures for Android, iOS, and macOS companion apps
frontend-design
84.5kCreate distinctive, production-grade frontend interfaces with high design quality. Use this skill when the user asks to build web components, pages, or applications. Generates creative, polished code that avoids generic AI aesthetics.
openai-whisper-api
341.2kTranscribe audio via OpenAI Audio Transcriptions API (Whisper).
commit-push-pr
84.5kCommit, push, and open a PR
