SV2
Support Vector Structural Variation Genotyper
Install / Use
/learn @dantaki/SV2README

Support Vector Structural Variation Genotyper
A genotyper for the rest of us
![]()
Danny Antaki, William M Brandler, Jonathan Sebat; SV<sup>2</sup>: Accurate Structural Variation Genotyping and De Novo Mutation Detection from Whole Genomes, Bioinformatics, , btx813, https://doi.org/10.1093/bioinformatics/btx813
SV<sup>2</sup> filters and integrates structural variants from multiple calling algorithms. Given multiple samples, SV<sup>2</sup> creates a genotype matrix. SV<sup>2</sup> also provides annotations for genes, repeat elements, and common SVs for filtering post-genotyping.
Table of Contents
- User Guide
- Getting Started
- Input
- Output
- Training
- Performance
- Requirements
- Citing SV<sup>2</sup>
- Troubleshooting
- License
- Contact
SV<sup>2</sup> (support-vector structural-variant genotyper) is a machine learning algorithm for genotyping deletions and duplications from paired-end whole genome sequencing data. SV<sup>2</sup> can rapidly integrate variant calls from multiple SV discovery algorithms into a unified callset with high genotyping accuracy and detection of de novo mutations.

User Guide
Tutorial
Getting Started
:one: Installation
Install with pip Recommended
$ pip install sv2
:two: Configure SV<sup>2</sup>
# download required resource files
$ sv2 -download
# define fasta locations
$ sv2 -hg19 /full/path/to/hg19.fa [-hg38 hg38.fa -mm10 mm10.fa]
:three: Run SV<sup>2</sup>
$ sv2 -i in.bam -v sv.vcf -snv in.vcf.gz -p in.ped
:notebook: SV<sup>2</sup> Options Documentation
Input
SV<sup>2</sup> requires BAM/CRAM files, SVs to genotype, SNV VCF files, and PED files.
$sv2 -i <in.bam ...> -v <sv.vcf ...> -b <sv.bed ...> -snv <snv.vcf.gz ...> -p <fam.ped ...>
Documentation for required inputs:
Output
Output is generated in the current working directory by default.
$ sv2 ... -O <output path prefix>
-
sv2_preprocessing/contains preprocessing output -
sv2_features/feature extraction output -
sv2_genotypes/genotype output
Output VCF contains annotations for genes and filters
-
SV<sup>2</sup> can merge divergent breakpoints. By default this option is off.
Training
Advanced users can retrain SV<sup>2</sup> genotyping classifiers with the original or custom training set.
:notebook: Training Documentation
Performance
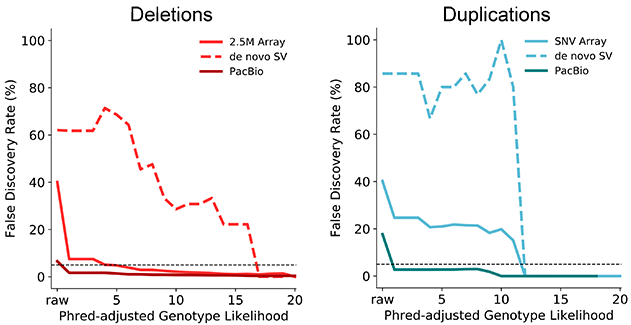
SV<sup>2</sup> estimated runtime performance. Details

SV<sup>2</sup> performance was determined with independent evaluation cohorts. Genotypes were validated with Illumina 2.5M microarrays and PacBio single molecule sequencing.

Performance of de novo mutations
Please refer to the publication for performance details.
Requirements
:notebook: Usage Documentation
Citing SV<sup>2</sup>
Danny Antaki, William M Brandler, Jonathan Sebat; SV<sup>2</sup>: Accurate Structural Variation Genotyping and De Novo Mutation Detection from Whole Genomes, Bioinformatics, , btx813, https://doi.org/10.1093/bioinformatics/btx813
Troubleshooting
License
MIT License
Copyright (c) 2017 Danny Antaki
Permission is hereby granted, free of charge, to any person obtaining a copy of this software and associated documentation files (the "Software"), to deal in the Software without restriction, including without limitation the rights to use, copy, modify, merge, publish, distribute, sublicense, and/or sell copies of the Software, and to permit persons to whom the Software is furnished to do so, subject to the following conditions:
The above copyright notice and this permission notice shall be included in all copies or substantial portions of the Software.
THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
Contact
:mailbox: dantaki@ucsd.edu :metal:

{kind=link}