Kneaddata
Quality control tool on metagenomic and metatranscriptomic sequencing data, especially data from microbiome experiments.
Install / Use
/learn @biobakery/KneaddataREADME
ATTENTION
Before opening a new issue here, please check the appropriate help channel on the KneadData bioBakery Support Forum and consider opening or commenting on a thread there.
For additional information, visit the KneadData Tutorial.
KneadData User Manual
KneadData is a tool designed to perform quality control on metagenomic and metatranscriptomic sequencing data, especially data from microbiome experiments. In these experiments, samples are typically taken from a host in hopes of learning something about the microbial community on the host. However, sequencing data from such experiments will often contain a high ratio of host to bacterial reads. This tool aims to perform principled in silico separation of bacterial reads from these "contaminant" reads, be they from the host, from bacterial 16S sequences, or other user-defined sources. Additionally, KneadData can be used for other filtering tasks. For example, if one is trying to clean data derived from a human sequencing experiment, KneadData can be used to separate the human and the non-human reads.
If you use the KneadData software, please cite our manuscript: TBA
Contents
Requirements
- Trimmomatic (version == 0.33) (automatically installed)
- Bowtie2 (version >= 2.2) (automatically installed)
- Python (version >= 2.7)
- Java Runtime Environment
- TRF (optional)
- Fastqc (optional)
- SAMTools (only required if input file is in BAM format)
- Memory (>= 4 Gb if using Bowtie2, >= 8 Gb if using BMTagger)
- Operating system (Linux or Mac)
Optionally, BMTagger can be used instead of Bowtie2.
The executables for the required software packages should be installed in your $PATH. Alternatively, you can provide the location of the Bowtie2 install ($BOWTIE2_DIR) with the following KneadData option “--bowtie2 $BOWTIE2_DIR”.
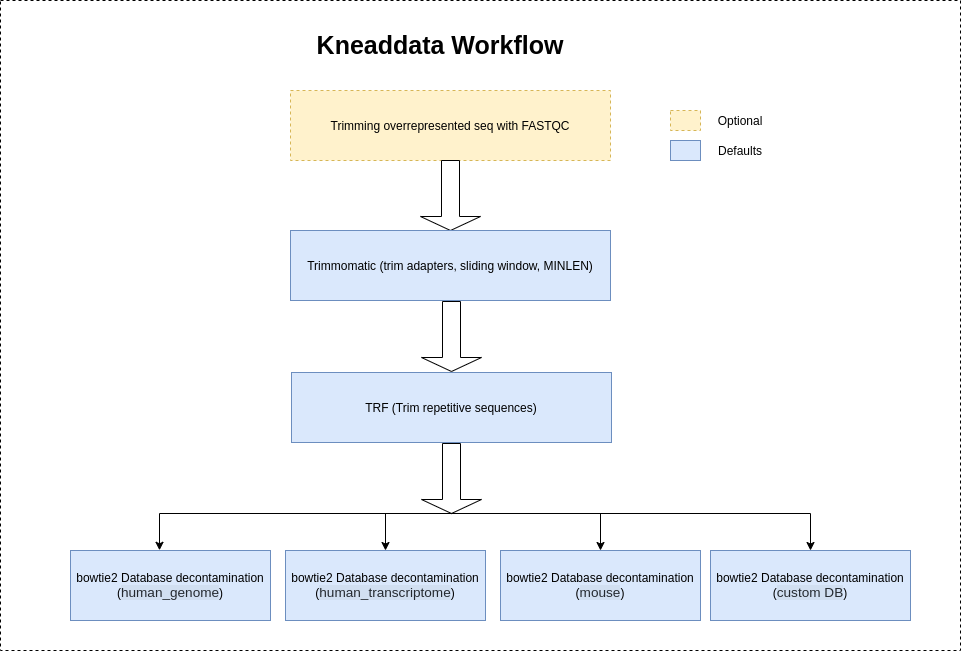
Installation
Before installing KneadData, please install the Java Runtime Environment (JRE). First download the JRE for your platform. Then follow the instructions for your platform: Linux 64-bit or Mac OS. At the end of the installation, add the location of the java executable to your $PATH.
Download KneadData
You can download the latest KneadData release or the development version. The source contains example files. If installing with pip, it is optional to first download the KneadData source.
Option 1: Latest Release (Recommended)
- Download kneaddata.tar.gz and unpack the latest release of KneadData.
Option 2: Development Version
-
Create a clone of the repository:
$ git clone https://github.com/biobakery/kneaddata.gitNote: Creating a clone of the repository requires Git to be installed.
Install KneadData
Install with pip
$ pip install kneaddata- This command will automatically install Trimmomatic and Bowtie2. To bypass the install of dependencies, add the option "--install-option='--bypass-dependencies-install'".
- If you do not have write permissions to '/usr/lib/', then add the option "--user" to the install command. This will install the python package into subdirectories of '$HOME/.local'. Please note when using the "--user" install option on some platforms, you might need to add '$HOME/.local/bin/' to your $PATH as it might not be included by default. You will know if it needs to be added if you see the following message
kneaddata: command not foundwhen trying to run KneadData after installing with the "--user" option.
Install from source
- Follow the instructions to download KneadData
- Move to the KneadData source directory:
$ cd kneaddata - Install KneadData
$ python setup.py install- This command will automatically install Trimmomatic and Bowtie2. To bypass the install of dependencies, add the option "--bypass-dependencies-install".
- If you do not have write permissions to '/usr/lib/', then add the option "--user" to the install command. This will install the python package into subdirectories of '$HOME/.local'. Please note when using the "--user" install option on some platforms, you might need to add '$HOME/.local/bin/' to your $PATH as it might not be included by default. You will know if it needs to be added if you see the following message
kneaddata: command not foundwhen trying to run KneadData after installing with the "--user" option.
Download the database
It is recommended that you download the human (Homo_sapiens_hg39_T2T_Bowtie2_v0.1.tar.gz - source ) reference database (approx. size = 3.6 GB). However, this step is not required if you are using your own custom reference database or if you will not be running with a reference database.
$ kneaddata_database --download human_genome bowtie2 $DIR- When running this command, $DIR should be replaced with the full path to the directory you have selected to store the database.
If you are running with bmtagger instead of bowtie2, then download the bmtagger database instead of the bowtie2 database with the following command.
$ kneaddata_database --download human_genome bmtagger $DIR- When running this command, $DIR should be replaced with the full path to the directory you have selected to store the database.
The human transcriptome (hg38) reference database is also available for download (approx. size = 254 MB).
$ kneaddata_database --download human_transcriptome bowtie2 $DIR
The SILVA Ribosomal RNA reference database is also available for download (approx. size = 11 GB).
$ kneaddata_database --download ribosomal_RNA bowtie2 $DIR
The mouse (C57BL) reference database is also available for download (approx. size = 3 GB).
$ kneaddata_database --download mouse_C57BL bowtie2 $DIR
The dog reference database (German Shepherd dog assembly) is also available for download (approximate size = ~2.5 Gb). This database is based on the genomic DNA sequences for the Canis lupus familiaris assembly version UU_Cfam_GSD_1.0 (accession GCF_011100685.1). This file includes the nucleotide sequences of the assembled chromosomes and unplaced scaffolds.
$ kneaddata_database --download dog_genome bowtie2 $DIR
The dog reference database (domestic dog) is also available for download (approx. size = 1.4 GB). This database is based on the genomic DNA sequences for the Canis familiaris (domestic dog) assembly version ROS_Cfam_1.0. This file includes the nucleotide sequences of the assembled chromosomes and unplaced scaffolds.
$ wget https://huttenhower.sph.harvard.edu/kneadData_databases/dog_genome.tar.gz
The cat reference database is available for download (approx. size = 3.7 GB). This database is based on the genomic DNA sequences for the Felis catus (domestic cat) This link includes the nucleotide sequences of the assembled chromosomes and unplaced scaffolds.
$ kneaddata_database --download cat_genome bowtie2 $DIR
Create a Custom Database
A reference database can be downloaded to use when running KneadData. Alternatively, you can create your own custom reference database.
Select Reference Sequences
First you must select reference sequences for the contamination you are trying to remove. Say you wish to filter reads from a particular "host." Broadly defined, the host can be an organism, or a set of organisms, or just a set of sequences. Then, you simply must generate a reference database for KneadData from a FASTA file containing these sequences. Usually, researchers want to remove reads from the human genome, the human transcriptome, or ribosomal RNA. You can access some of these FASTA files using the resources below:
- Ribosomal RNA: Silva provides a comprehensive database for ribosomal RNA sequences spanning all three domains of life (Bacteria, Archaea, and *
