Fastv
An ultra-fast tool for identification of SARS-CoV-2 and other microbes from sequencing data. This tool can be used to detect viral infectious diseases, like COVID-19.
Install / Use
/learn @OpenGene/FastvREADME
fastv
fastv is an ultra-fast tool for identification of SARS-CoV-2 and other microbes from sequencing data. It detects microbial sequences from FASTQ data, generates JSON reports and visualizes the result in HTML reports. This tool can be used to detect viral infectious diseases, like COVID-19. This tool supports both short reads (Illumina, BGI, etc.) and long reads (ONT, PacBio, etc.)
- Examples
- How it works?
- Understand the input
- Understand the output
- Download or build fastv
- Screenshot
- Options
- Citation
- Tutorials
- ---- mNGS data analysis
- ---- SARS-CoV-2 identification
- ---- influenza A virus subtyping
take a quick glance of the informative report
- Sample HTML report (Illumina): http://opengene.org/fastv/fastv.html
- Sample HTML report (ONT): http://opengene.org/fastv/ont.html
- Sample JSON report: http://opengene.org/fastv/fastv.json
quick example for SARS-CoV-2 identification
- download FASTQ file for testing: http://opengene.org/fastv/testdata.fq.gz
- get fastv and use following command for testing:
# make sure that SARS-CoV-2.kmer.fa and SARS-CoV-2.genomes.fa are in the ./data folder
./fastv -i testdata.fq.gz
how it works?
fastv accepts FASTQ files as input, and then:
- performs data QC and quality filtering as
fastpdoes (cut adapters, remove low quality reads, correct wrong bases). - scans the clean data to collect the sequences that containing any unique k-mer, or can be mapped to any reference microbial genomes.
- makes statistics, visualizes the result in HTML format, and output the results in JSON format.
- outputs the on-target sequencing reads so that they can be analyzed by downstream tools.
understand the input
fastv accepts following files as input:
FASTQfile (required) to be scanned, can be single-end (-i) or paired-end (-iand-I), can be short reads (Illumina, MGI, etc.) or long reads (PacBio, ONT, etc.)genomesfile (optional): a FASTA file containing one or many reference genomes of the target microorganism (-g).k-merfile (optional): a FASTA file containing the UNIQUE k-mer of the target microbial genomes (-k).k-mer collectionfile (optional): a FASTA containing the unique k-mers of many microorganisms (-c). See an example: http://opengene.org/kmer_collection.fasta
If none of (k-mer, k-mer collection, genomes) files is specified, fastv will try to load the SARS-CoV-2 Genomes/k-mer files in the data folder to detect SARS-CoV-2 sequences.
Besides the HTML/JSON reports, fastv also can output the sequence reads that contains any unique k-mer or can be mapped to any of the target reference genomes. The output data:
- is in FASTQ format
- is clean data after quality filtering
- the file names can be specified by
-ofor SE data, or-oand-Ofor PE data.
get the pre-built k-mer file, genomes file or k-mer collection file for viruses
- You can download
k-merfiles andgenomesfiles of viruses from http://opengene.org/uniquekmer/viral/index.html. This is generated by extracting unique k-mers for all genomes in a big FASTA (http://opengene.org/viral.genomic.fasta), which contains all NCBI complete RefSeq release of viral sequences that can be found from https://ftp.ncbi.nlm.nih.gov/refseq/release/viral/. The k-mers that can be mapped to human reference genome (GRCh38) withedit_distance <= 3have already been filtered out. - You can download the
k-mer collectionfile for viral genomes from: http://opengene.org/viral.kc.fasta.gz
get the pre-built k-mer file, genomes file or k-mer collection file for viruses and human microorganisms
- You can download
k-merfiles andgenomesfiles of viruses from http://opengene.org/uniquekmer/microbial/index.html. This is generated by extracting unique k-mers for all genomes in a big FASTA (http://opengene.org/microbial.genomic.fasta), which contains genomes for the viruses above and common human microorganisms. The k-mers that can be mapped to human reference genome (GRCh38) withedit_distance <= 3have already been filtered out. - You can download the
k-mer collectionfile for viral and microbial genomes from: http://opengene.org/microbial.kc.fasta.gz - you can get the
k-merfile andgenomesfile for SARS-CoV-2 bygit clone https://github.com/OpenGene/fastv.git. If you don't use git, you can also download these two files from http://opengene.org/fastv/SARS-CoV-2.kmer.fa and http://opengene.org/fastv/SARS-CoV-2.genomes.fa
If you want to generate your own unique k-mer files and k-mer collection files, please use UniqueKMER: https://github.com/OpenGene/UniqueKMER
understand the output
fastv outputs reports in HTML and JSON formats.
- Sample HTML report (Illumina): http://opengene.org/fastv/fastv.html
- Sample JSON report: http://opengene.org/fastv/fastv.json
- If the
k-merfile is specified, there will be aPOSITIVEorNEGATIVEresult, which is determined by comparing the mean depth of the k-mer keys to the threshold (--positive_threshold).
Besides the HTML/JSON reports, fastv also can output the sequence reads that contains any unique k-mer or can be mapped to any of the target reference genomes. The output data:
- is in FASTQ format
- is clean data after quality filtering
- the file names can be specified by
-ofor SE data, or-oand-Ofor PE data.
get fastv
download binary
This binary is only for Linux systems: http://opengene.org/fastv/fastv
# this binary was compiled on CentOS, and tested on CentOS/Ubuntu
wget http://opengene.org/fastv/fastv
chmod a+x ./fastv
or compile from source
# step 1: get the code
git clone https://github.com/OpenGene/fastv.git
# step 2: build
cd fastv
make
# step 3: install it to system if you have a sudo permission
make install
screenshot
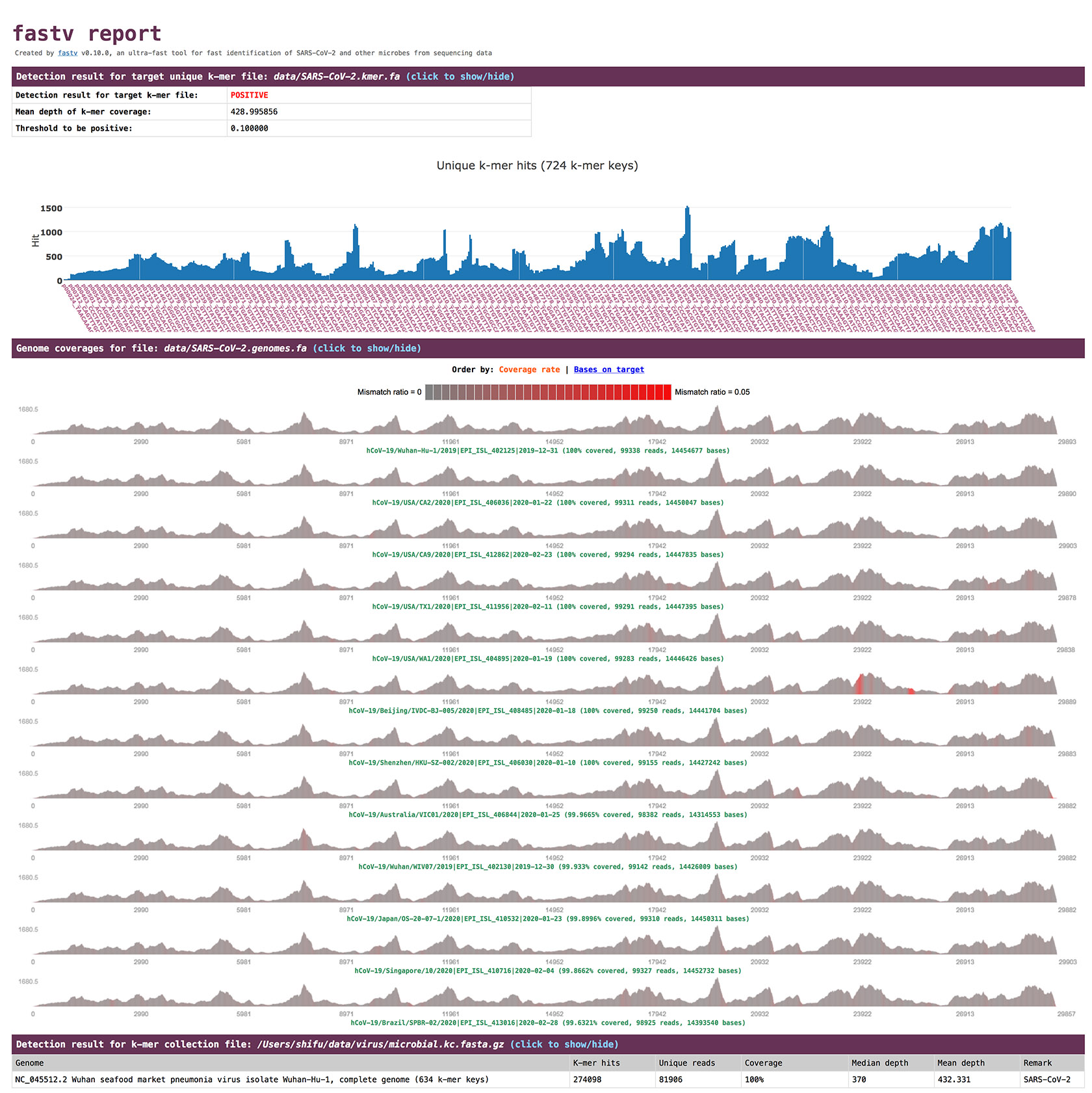
options
Key options:
-i, --in1 read1 input file name (string [=])
-I, --in2 read2 input file name (string [=])
-o, --out1 file name to store read1 with on-target sequences (string [=])
-O, --out2 file name to store read2 with on-target sequences (string [=])
-c, --kmer_collection the unique k-mer collection file in fasta format, see an example: http://opengene.org/kmer_collection.fasta (string [=])
-k, --kmer the unique k-mer file of the detection target in fasta format. data/SARS-CoV-2.kmer.fa will be used if none of k-mer/Genomes/k-mer_Collection file is specified (string [=])
-g, --genomes the genomes file of the detection target in fasta format. data/SARS-CoV-2.genomes.fa will be used if none of k-mer/Genomes/k-mer_Collection file is specified (string [=])
-p, --positive_threshold the data is considered as POSITIVE, when its mean coverage of unique kmer >= positive_threshold (0.001 ~ 100). 0.1 by default. (float [=0.1])
-d, --depth_threshold For coverage calculation. A region is considered covered when its mean depth >= depth_threshold (0.001 ~ 1000). 1.0 by default. (float [=1])
-E, --ed_threshold If the edit distance of a sequence and a genome region is <=ed_threshold, then consider it a match (0 ~ 50). 8 by default. (int [=8])
--long_read_threshold A read will be considered as long read if its length >= long_read_threshold (100 ~ 10000). 200 by default. (int [=200])
--read_segment_len A long read will be splitted to read segments, with each <= read_segment_len (50 ~ 5000, should be < long_read_threshold). 100 by default. (int [=100])
--bin_size For coverage calculation. The genome is splitted to many bins, with each bin has a length of bin_size (1 ~ 100000), default 0 means adaptive. (int [=0])
--kc_coverage_threshold For each genome in the k-mer collection FASTA, report it when its coverage > kc_coverage_threshold. Default is 0.01. (double [=0.01])
--kc_high_confidence_coverage_threshold For each genome in the k-mer collection FASTA, report it as high confidence when its coverage > kc_high_confidence_coverage_threshold. Default is 0.9. (double [=0.9])
--kc_high_confidence_median_hit_threshold For each genome in the k-mer collection FASTA, report it as high confidence when its median hits > kc_high_confidence_median_hit_threshold. Default is 5. (int [=5])
-j, --json the json format report file name (string [=fastv.json])
-h, --html the html format report file name (string [=fastv.html])
-R, --report_title should be quoted with ' or ", default is "fastv report" (string [=fastv report])
-w, --thread worker thread number, default is 4 (int [=4])
Other I/O options:
-6, --phred64 indicate the input is using phred64 scoring (it'll be converted to phred33, so the output will still be phred33)
-z, --compression compression level for gzip output (1 ~ 9). 1 is fastest, 9 is smallest, default is 4. (int [=4])
--stdin input from STDIN. If the STDIN is interleaved paired-end FASTQ, please also add --interleaved_in.
--stdout stream passing-filters reads to STDOUT. This option will result in interleaved FASTQ ou
Related Skills
node-connect
330.3kDiagnose OpenClaw node connection and pairing failures for Android, iOS, and macOS companion apps
frontend-design
81.3kCreate distinctive, production-grade frontend interfaces with high design quality. Use this skill when the user asks to build web components, pages, or applications. Generates creative, polished code that avoids generic AI aesthetics.
openai-whisper-api
330.3kTranscribe audio via OpenAI Audio Transcriptions API (Whisper).
commit-push-pr
81.3kCommit, push, and open a PR
