GenoPHI
Python package for phage-host interaction prediction
Install / Use
/learn @Noonanav/GenoPHIREADME
GenoPHI
(jee-no-fee)
Genotype-to-Phenotype Phage-Host Interaction Prediction
GenoPHI is a Python package for machine learning-based prediction of genotype-phenotype relationships using whole-genome sequence data. Originally designed for phage-host interaction prediction, GenoPHI supports both binary interaction prediction and regression tasks for any microbial phenotype. The package implements protein family-based and k-mer-based approaches to extract genomic features from amino acid sequences and predict phenotypes using CatBoost gradient boosting models.
![]()
![]()
![]()
Workflow Overview
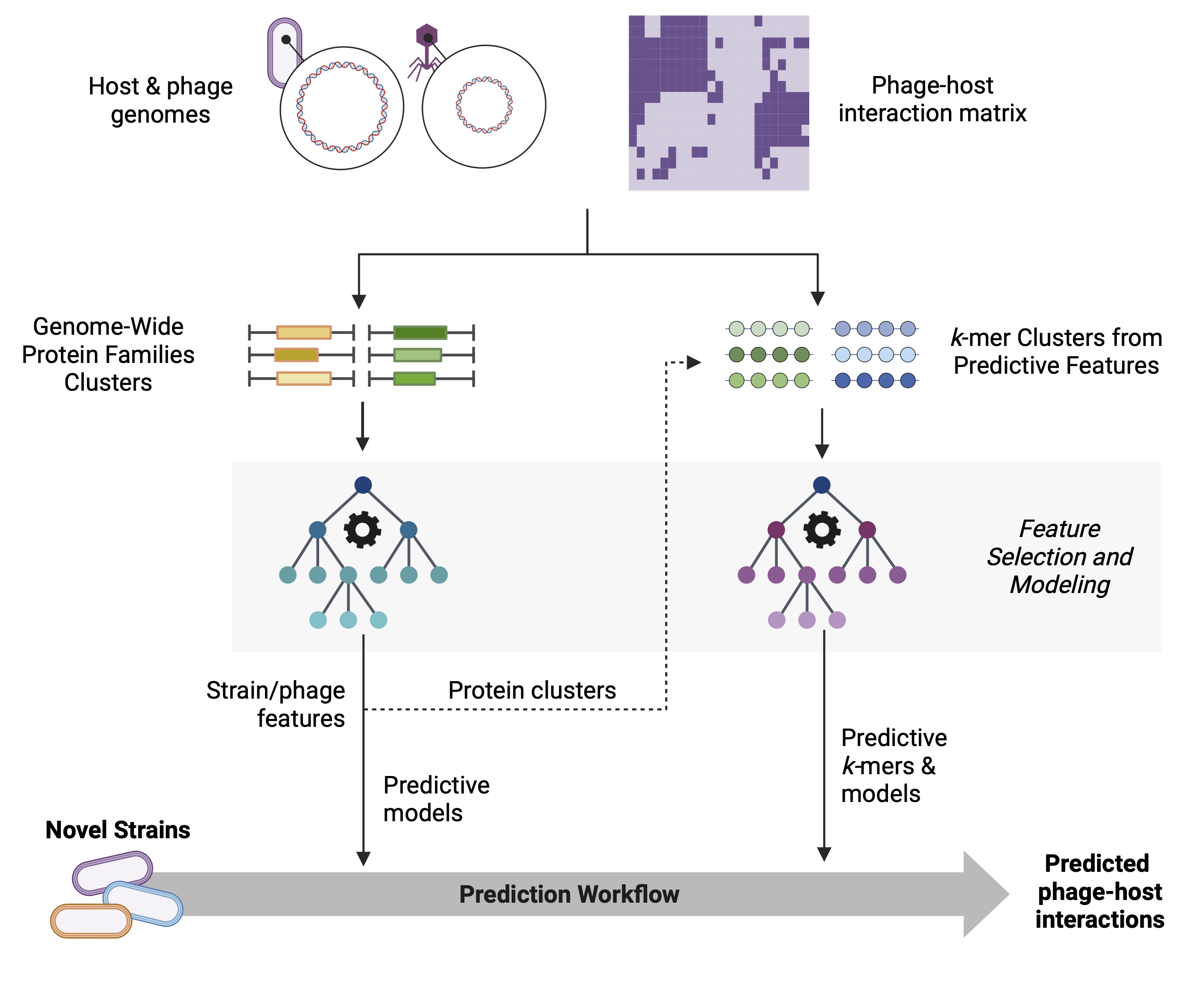 Figure 1: GenoPHI workflow schematic showing the main analysis pipelines: Protein family-based workflow, K-mer-based workflow, and Predictive protein k-mer workflow. Each pathway includes feature extraction, selection, model training, and prediction steps.
Figure 1: GenoPHI workflow schematic showing the main analysis pipelines: Protein family-based workflow, K-mer-based workflow, and Predictive protein k-mer workflow. Each pathway includes feature extraction, selection, model training, and prediction steps.
Table of Contents
- Features
- Installation
- Quick Start
- Usage
- Input Data Formats
- Feature Selection Methods
- Performance Metrics
- Output Directory Structure
- Python API
- Advanced Usage
- Troubleshooting
- Testing
- Best Practices
- Citation
- Contributing
- License
- Support
- Acknowledgments
Features
Protein Family-Based Analysis
- MMSeqs2 Clustering: Cluster protein sequences into protein families based on sequence similarity
- Feature Table Generation: Create presence-absence matrices of protein families across genomes and consolidate into predictive features based on co-occurence across genomes
- Feature Selection: Identify predictive protein families (multiple available methods: RFE, SHAP, SHAP-RFE, ANOVA, Chi-squared, Lasso)
- Model Training: Train CatBoost models with hyperparameter optimization
- Phenotype Prediction: Predict interactions, resistance, or other phenotypes for new genomes
- Feature Annotation: Identify predictive protein sequences from predictive features
K-mer-Based Analysis
- K-mer Feature Extraction: Generate k-mer features from protein sequences with or without gene family context
- Predictive K-mer Workflow: Extract k-mers specifically from predictive protein families identified in protein family analysis
- Feature Selection & Modeling: Apply same robust feature selection and modeling pipelines
- Flexible K-mer Lengths: Support for single k-mer length or ranges (e.g., 3-6)
Application Modes
- Phage-Host Interaction Prediction: Binary prediction of infection outcomes between phages and bacterial strains
- Single-Strain Phenotype Prediction: Predict strain-level phenotypes (e.g., antibiotic resistance, growth rate) without requiring phage data
- Regression Tasks: Predict continuous phenotypes (e.g., infection efficiency, metabolic rates)
- General Feature-Based Modeling: Use any feature table with a phenotype column for custom applications
Advanced Capabilities
- Dynamic Feature Weighting: Account for feature frequency distributions to handle imbalanced features
- Clustering-Based Selection: Use HDBSCAN or hierarchical clustering for intelligent feature grouping
- Multiple Feature Selection Methods: RFE, SHAP-RFE, SelectKBest, Chi-squared, Lasso, SHAP
- Comprehensive Performance Metrics: AUC-ROC, Precision-Recall, MCC, F1-score, Accuracy
- SHAP Interpretability: Feature importance analysis and visualization for model explainability
- Bootstrapping Support: Robust model evaluation with multiple train-test splits
Installation
System Requirements
Minimum Requirements:
- Python 3.8 or higher
- 8 GB RAM
- 4 CPU cores
- 10 GB free disk space
Recommended for Large Datasets:
- Python 3.10+
- 32+ GB RAM
- 8+ CPU cores
- 50+ GB free disk space (depending on dataset size)
Tested Operating Systems:
- Linux (Ubuntu 20.04+, CentOS 7+)
- macOS (Sonoma 14+, Apple Silicon)
Virtual Environment (Recommended)
Create and activate a conda environment:
conda create -n genophi python=3.10
conda activate genophi
Install GenoPHI
From PyPI (Recommended):
pip install genophi
From GitHub (Development):
git clone https://github.com/Noonanav/GenoPHI.git
cd GenoPHI
pip install -e .
For development with optional dependencies:
pip install -e ".[dev]"
Install MMseqs2
External Dependency: GenoPHI requires MMseqs2 for protein sequence clustering and assignment.
Install via conda/mamba:
conda install -c bioconda mmseqs2
# or
mamba install -c bioconda mmseqs2
For other installation methods, see the MMSeqs2 Wiki.
Verify Installation
Test that GenoPHI is properly installed:
# Check GenoPHI version
genophi --version
# Verify MMseqs2 is accessible
mmseqs version
# Run basic help command
genophi --help
Typical Install Time
Full installation (conda environment + GenoPHI + MMseqs2) takes approximately 2-3 minutes on a standard desktop computer (tested on a MacBook Pro M2, 16 GB RAM, macOS Sonoma 14.3).
Demo
A small test dataset is included in the repository for demonstrating the software. To run the demo:
git clone https://github.com/Noonanav/GenoPHI.git
cd GenoPHI
genophi protein-family-workflow \
--input_path_strain data/test_data/strain_AAs/ \
--input_path_phage data/test_data/phage_AAs/ \
--phenotype_matrix data/test_data/ecoli_test_interaction_matrix.csv \
--output_dir demo_output/ \
--threads 4 \
--num_features 50 \
--num_runs_fs 5 \
--num_runs_modeling 10 \
--method rfe \
--filter_type strain
Test dataset: 25 E. coli strains and 25 phages with a binary interaction matrix.
Expected output: A demo_output/ directory containing MMseqs2 clustering results, feature selection outputs, trained models, performance metrics, and a workflow summary report. See Output Directory Structure for details.
Expected run time: ~25 minutes on a standard desktop computer (MacBook Pro M2, 16 GB RAM).
Quick Start
GenoPHI provides a unified command-line interface accessible through the genophi command:
# View available commands
genophi --help
# Get help for a specific command
genophi protein-family-workflow --help
Recommended Default Run
For most phage-host interaction prediction tasks, use these recommended settings:
genophi protein-family-workflow \
--input_path_strain strain_fastas/ \
--input_path_phage phage_fastas/ \
--phenotype_matrix interactions.csv \
--output_dir results/ \
--threads 8 \
--num_features 100 \
--num_runs_fs 25 \
--num_runs_modeling 50 \
--method rfe \
--use_clustering \
--cluster_method hierarchical \
--n_clusters 20 \
--filter_type strain \
--use_shap
Key Parameters Explained:
--num_features 100: Select top 100 features (adjust based on dataset size)--num_runs_fs 25: 25 iterations for robust feature selection--num_runs_modeling 50: 50 modeling runs for reliable performance estimates--method rfe: Recursive Feature Elimination (balanced performance)--use_clustering: Enable sample clustering-aware filtering--filter_type strain: Critical for phage-host prediction - Ensures train/test splits separate by strain so the model learns to predict on new strains it hasn't seen before--use_shap: Generate SHAP plots and feature importance analysis for model interpretability
Note: For phage-host interaction prediction, --filter_type strain is strongly recommended. This controls how train/test splits are made during feature selection and modeling, ensuring the model never sees the same strain in both training and testing. This forces the model to learn generalizable patterns rather than memorizing specific strain characteristics.
For single-strain phenotypes (no phage data):
genophi protein-family-workflow \
--input_path_strain strain_fastas/ \
--phenotype_matrix phenotypes.csv \
--output_dir results/ \
--threads 8 \
--sample_column strain \
--phenotype_column resistance \
--filter_type none
Usage
CLI Commands Overview
GenoPHI provides the following main commands:
| Command | Description |
|---------|-------------|
| protein-family-workflow | Recommended basic workflow: Complete protein family-based workflow |
| full-workflow | Protein families → k-mers from predictive proteins |
| kmer-workflow | Complete k-mer-based workflow from all proteins |
| cluster | Generate protein family clusters and feature tables |
| select-features | Perform feature selection on any feature table |
| train | Train predictive models on selected features |
| predict | Predict phenotypes using trained models |
| select-and-train | Feature selection + modeling from any feature table |
| assign-features | Assign features to new genomes |
| assign-predict | Assign features and predict (protein families) |
| annotate | Annotate predictive features with functional info |
| kmer-assign-features | Assign k-mer features to new genomes |
| `kmer-assign-p
Related Skills
node-connect
354.5kDiagnose OpenClaw node connection and pairing failures for Android, iOS, and macOS companion apps
frontend-design
112.4kCreate distinctive, production-grade frontend interfaces with high design quality. Use this skill when the user asks to build web components, pages, or applications. Generates creative, polished code that avoids generic AI aesthetics.
openai-whisper-api
354.5kTranscribe audio via OpenAI Audio Transcriptions API (Whisper).
qqbot-media
354.5kQQBot 富媒体收发能力。使用 <qqmedia> 标签,系统根据文件扩展名自动识别类型(图片/语音/视频/文件)。
