PHap
A haplotype-resolved and telomere-to-telomere genome assembly pipeline (PHap) tailored for autopolyploids, relying solely on common sequencing data including long-reads and Hi-C.
Install / Use
/learn @JiaoLab2021/PHapREADME
PHap
A haplotype-resolved and telomere-to-telomere genome assembly pipeline (PHap) tailored for autopolyploids, relying solely on common sequencing data including long-reads and Hi-C.
Overview
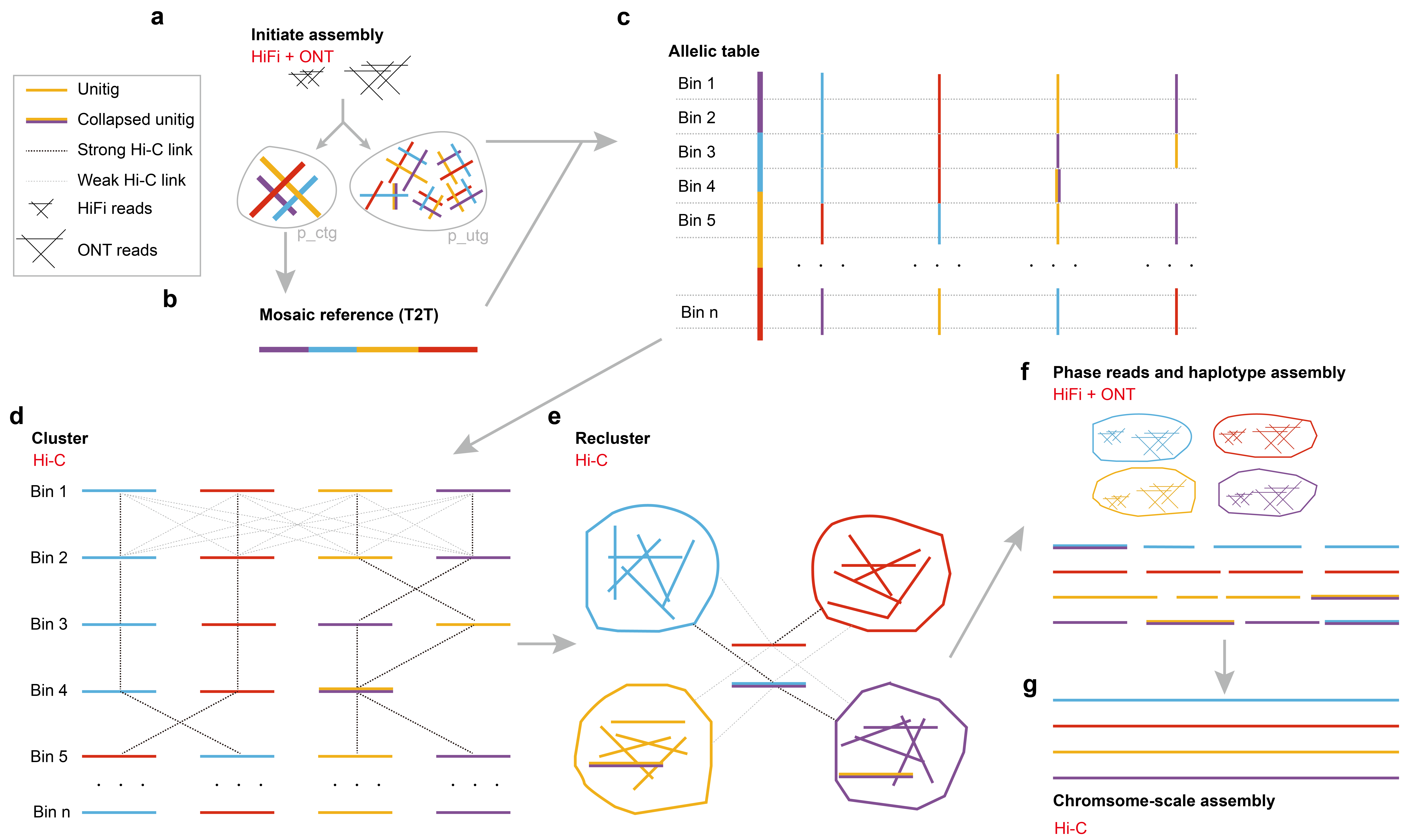
- Initial assembly. A primary contig assembly (p_ctg) and a phased unitig assembly (p_utg) are assembled using hifiasm with the long-read sequencing data including PacBio HiFi and Oxford Nanopore Ultra-Long sequencings.
- mT2T assembly. A mosaic T2T (mT2T) reference is assembled based on the all-vs-all alignments of the p_ctg assembly contigs.
- Allelic unitig table construction. All unitigs of the p_utg assembly are aligned to the mT2T reference to build an allelic table.
- Unitig clustering. Unitigs in the allelic table are clustered according to the strength of Hi-C signals and guided by alignments against mT2T.
- Unitig re-clustering. Re-clustering for each of remaining unitigs based on their relative intensity of Hi-C interaction against each group.
- Read phasing and de novo assembly. Long reads are mapped to the p_utg unitigs, assigned into haplotypes, and de novo assembled independently for each haplotype.
- Chromosome-scale assembly. Contigs of each haplotype assembly are scaffolded with Hi-C data and followed by gap filling and polishing with long reads.
System Requirements
- All scripts and analyses were developed and tested on Linux operating system environment.
- Several essential tools were required:
Installation
- Download PHap
$ git clone git@github.com:JiaoLab2021/PHap.git
$ cd /path/to/PHap/
$ chmod +x PHap.py
Usage
- View the help document of PHap
$ /path/to/PHap/PHap.py -h
Usage: phap [command] <parameters>
Command Description
-------- ------------------------------------------------------------------------
mt2t Generate a mosaic telomere-to-telomere genome using the p_ctg genome as
the reference. This is used for creating an allelic contig table to solve
the allelic conflict problem.
cluster Cluster contigs. First, extract contigs corresponding to each chromosome
based on mT2T alignment. Then, cluster the contigs for each haplotype of
each chromosome using Hi-C signals. Third, cluster the contigs not on mT2T
chromosomes to clustered group.
phase_reads Based on the clustering results, the corresponding reads of each haplotype
are extracted, then assembled and anchored separately.
Use phap [command] --help/-h to see detailed help for each individual command.
- View the help document of subcommands of PHap
$ /path/to/PHap/PHap.py mt2t -h
usage: Get mosaic T2T (mT2T) reference from primary contig assembly (p_ctg).
$ /path/to/PHap/PHap.py cluster -h
usage: Haplotype clustering of autopolyploid genome.
$ /path/to/PHap/PHap.py phase_reads -h
usage: Haplotype assembly and scaffolding of autopolyploid genome.
- For convenience, users can add
/path/to/PHap/PHap.pyto environment variables, such as:ln -s /path/to/PHap/PHap.py ~/.local/bin/phap, and then thephapcommand can be executed directly from any location in the terminal.
The pipeline for assembling a tetraploid potato genome
Please check the Pipeline.
Note
PHap is currently developed to do haplotype-resolved telomere-to-telomere (T2T) genome assembly in autotetraploid genomes, with a primary focus on crops such as potato (Solanum tuberosum). Theoretically, PHap can be easily extended to support other polyploid genome types, including triploid, hexaploid, and more complex genomes.
