NtEdit
✏️ Genome assembly polishing & SNV detection
Install / Use
/learn @BirolLab/NtEditREADME




Thank you for your

![]()
ntEdit
Fast, lightweight, scalable genome sequence polishing and SNV detection & annotation
2018-current
Contents
- Description
- Implementation and requirements
- Install
- Dependencies
- Documentation
- Citing ntEdit
- Credits
- How to run ntEdit
- Running ntEdit
- ntEdit polishing options
- Soft-mask option
- SNV mode
- VCF input option
- Test data
- Algorithm
- Output files
- License
Description <a name=description></a>
ntEdit is a fast and scalable genomics application for polishing genome sequence assembly drafts. It simplifies polishing, variant detection and "haploidization" of gene and genome sequences with its re-usable Bloom filter design. Although it was originally designed as a general-purpose polishing tool, initally aimed at improving genome sequences by fixing base mismatches and frame shift errors with the help of more base-accurate short sequencing reads, ntEdit can also be used for polishing with long reads and to "finish" genome sequence assembly projects (refer to <a href="https://github.com/birollab/goldPolish" target="_blank">GoldPolish</a> and the <a href="https://github.com/birollab/ntedit_sealer_protocol" target="_blank">ntedit+sealer genome assembly finishing protocol</a>, respectively).
We anticipate that ntEdit will find further applications in the rapid mapping of single nucleotide variants, as demonstrated below with the genome of SARS-CoV-2, the highly transmissible pathogenic coronavirus, and etiological agent of COVID-19. Additionally, for researchers delving into the intricate roots of genetic lineages within large cohort data, we encourage the utilization of <a href="https://github.com/birollab/ntroot" target="_blank">ntRoot, an ancestry prediction framework</a> built upon the ntEdit engine. It offers a comprehensive analysis of genetic heritage, employing sequence alignment-free algorithms to unveil ancestral connections and provide insights into genetic ancestry within diverse populations.
! NOTE: In v1.3.1 onwards, the parameter k is automatically detected from supplied Bloom filters
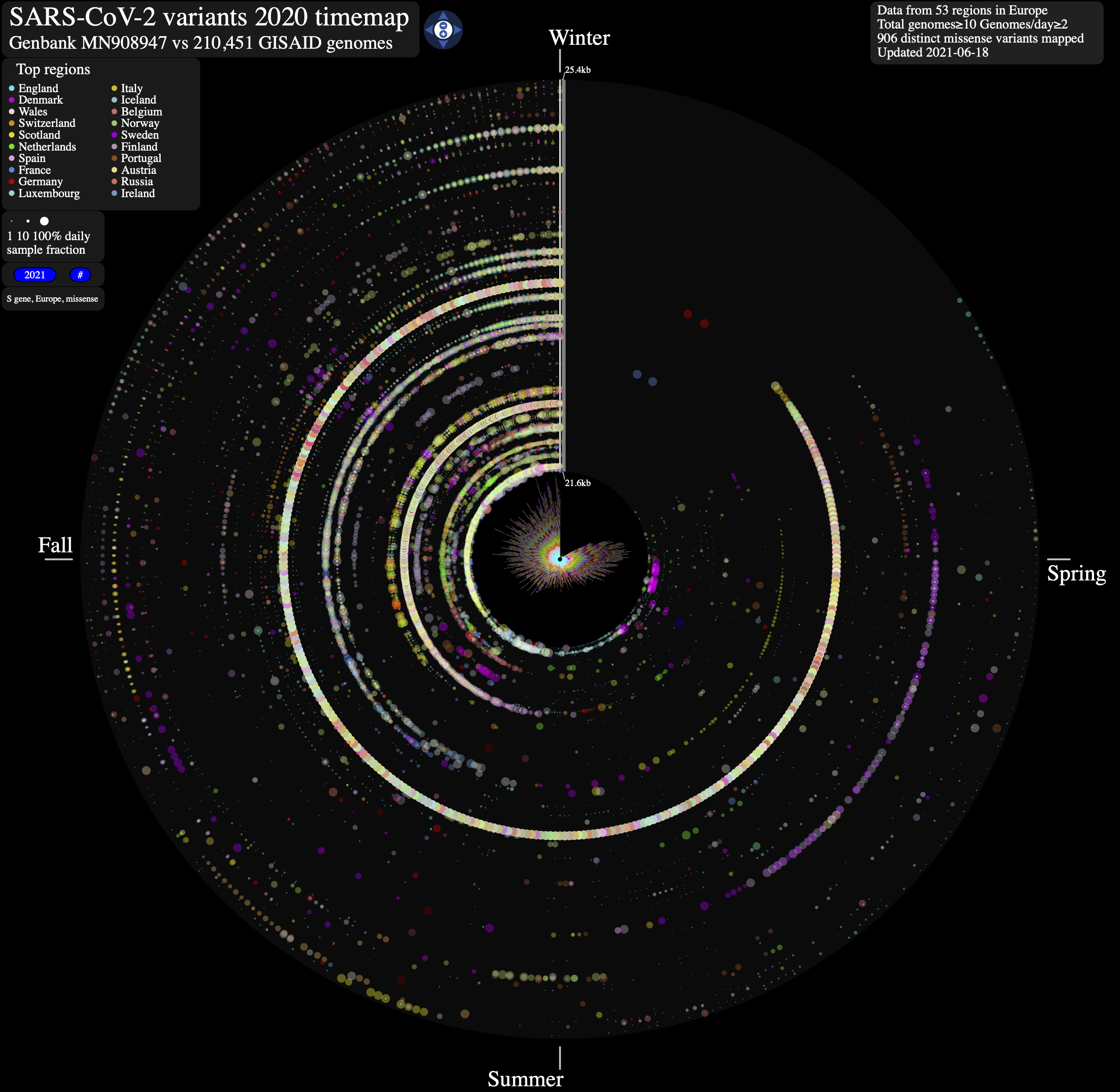 SARS-CoV-2 evolution in human hosts. ntEdit v1.3.4 was used to map nucleotide variation between the first published coronavirus isolate from Wuhan in early January 2020 and over 1,500,000 SARS-CoV-2 genomes sampled from around the globe during the COVID-19 pandemic. <a href="https://bcgsc.github.io/SARS2" target="_blank">Additional (& interactive) timemaps are available</a>.
SARS-CoV-2 evolution in human hosts. ntEdit v1.3.4 was used to map nucleotide variation between the first published coronavirus isolate from Wuhan in early January 2020 and over 1,500,000 SARS-CoV-2 genomes sampled from around the globe during the COVID-19 pandemic. <a href="https://bcgsc.github.io/SARS2" target="_blank">Additional (& interactive) timemaps are available</a>.
Implementation and requirements <a name=implementation></a>
ntEdit v1.2.0 and subsequent versions are written in C++.
(We compiled with gcc 5.5.0)
Install <a name=install></a>
Clone and enter the ntEdit directory.
<pre> git clone https://github.com/birollab/ntEdit.git cd ntEdit </pre>Compile ntEdit.
<pre> meson setup build --prefix=/path/to/ntedit/install/dir cd build ninja install </pre>Dependencies <a name=dependencies></a>
- ntStat (v1.0.0+, https://github.com/birollab/ntstat)
- BloomFilter utilities (provided in ./lib)
- kseq (provided in ./lib)
- meson
- ninja
- btllib
- snakemake
- python 3.9+
! NOTE: ntEdit v2.1.0+ IS ONLY compatible with ntStat release v1.0.0+
We recommend installing ntEdit and its dependencies, using conda:
<pre> conda install -c bioconda ntedit </pre>Documentation <a name=docs></a>
Refer to the README.md file on how to install and run ntEdit.
Our manuscript contains information about the software and its performance.
 This ISMB2019 poster contains additional information, benchmarks and results.
This ISMB2019 poster contains additional information, benchmarks and results.
Citing ntEdit <a name=citing></a>
Thank you for your and for using, developing and promoting this free software!
If you use ntEdit in your research, please cite:
ntEdit: scalable genome sequence polishing
<pre> ntEdit: scalable genome sequence polishing. Warren RL, Coombe L, Mohamadi H, Zhang J, Jaquish B, Isabel N, Jones SJM, Bousquet J, Bohlmann J, Birol I. Bioinformatics. 2019 Nov 1;35(21):4430-4432. doi: 10.1093/bioinformatics/btz400. </pre>The experimental data described in our paper can be downloaded from: http://www.bcgsc.ca/downloads/btl/ntedit/
Credits <a name=credits></a>
ntedit (concept, algorithm design and prototype): Rene Warren
nthash: Hamid Mohamadi, Parham Kazemi
ntstat: Parham Kazemi
C++ implementation: Jessica Zhang, Rene Warren, Johnathan Wong
Integration tests: Murathan T Goktas
ntEdit workflow: Johnathan Wong and Lauren Coombe
How to run ntEdit <a name=howto></a>
General ntEdit usage:
run-ntedit --help
usage: run-ntedit [-h] {polish,snv} ...
ntEdit: Fast, lightweight, scalable genome sequence polishing and SNV detection & annotation
positional arguments:
{polish,snv} ntEdit can be run in polishing or SNV modes.
polish Run ntEdit polishing
snv Run ntEdit SNV mode (Experimental)
optional arguments:
-h, --help show this help message and exit
Running in polishing mode <a name=run></a>
run-ntedit polish --help
usage: run-ntedit polish [-h] --draft DRAFT --reads READS [-i {0,1,2,3,4,5}] [-d {0,1,2,3,4,5,6,7,8,9,10}] [-x X] [--cap CAP] [-m {0,1,2}] [-a {0,1}] -k K
[-l L] [--cutoff CUTOFF] [--solid] [-t T] [-z Z] [-y Y] [-j J] [-X X] [-Y Y] [-v] [-V] [-n] [-f]
optional arguments:
-h, --help show this help message and exit
--draft DRAFT Draft genome assembly. Must be specified with exact FILE NAME. Ex: --draft myDraft.fa (FASTA, Multi-FASTA, and/or gzipped compatible),
REQUIRED
--reads READS Prefix of reads file(s). All files in the working directory with the specified prefix will be used for polishing (fastq, fasta, gz),
REQUIRED
-i {0,1,2,3,4,5} Maximum number of insertion bases to try, range 0-5, [default=5]
-d {0,1,2,3,4,5,6,7,8,9,10}
Maximum number of deletions bases to try, range 0-10, [default=5]
-x X k/x ratio for the number of k-mers that should be missing, [default=5.000]
--cap CAP Cap for the number of base insertions that can be made at one position[default=k*1.5]
-m {0,1,2} Mode of editing, range 0-2, [default=0] 0: best substitution, or first good indel 1: best substitution, or best indel 2: best edit
overall (suggestion that you reduce i and d for performance)
-a {0,1} Soft masks missing k-mer positions having no fix (1 = yes, default = 0, no)
-k K k-mer size, REQUIRED
-l L input VCF file with annotated variants (e.g., clinvar.vcf)
--cutoff CUTOFF The minimum coverage of k-mers in output Bloom filter [default=2, ignored if solid=True]
--solid Output the solid k-mers (non-erroneous k-mers), [default=False]
-t T Number of threads [default=4]
-z Z Minimum contig length [default=100]
-y Y k/y ratio for the number of edited k-mers that should be present, [default=9.000]
-j J controls size of k-mer subset. When checking subset of k-mers, check every jth k-mer [default=3]
-X X Ratio of number of k-mers in the k subset that should be missing in orderto attempt fix (higher=stringent) [default=0.5, if -Y is
specified]
-Y Y Ratio of number of k-mers in the k subset that should be present to accept an edit (higher=stringent) [default=0.5, if -X is specified]
-v Verbose mode, [default=False]
-V, --version show program's version number and exit
-n, --dry-run Print out the commands that will be executed
-f, --force Run all ntEdit steps, regardless of existing output files
Running ntEdit in SNV mode
run-ntedit snv --help
usage: run-ntedit snv [-h] [--reference REFERENCE] [--reads READS] [--genome GENOME [GENOME ...]] -k K [-l L] [--cutoff CUTOFF] [--solid] [-t T] [-z Z] [-y Y]
[-j J] [-X X] [-Y Y] [-v] [-V] [-n] [-f]
optional arguments:
-h, --help show this help message and exit
--reference REFERENCE
Reference genome assembly for SNV calling (FASTA, Multi-FASTA, and/or gzipped compatible), REQUIRED
